Wielding New Cancer Weapons Forged in Cell-Free DNA: A Wake Up Call For The Entire Biotech Industry
Advances in computational biology, precision medicine, artificial intelligence/machine learning, etc., are launching us into a world where strategic control points in drug discovery, pharmaceuticals, clinical trials, etc., will never be the same.

Image credit: Adrienn Harto
Drugs that were once nearly impossible to derive will become facile to make. Every protein in the cell will likely have a corresponding drug targeting it. Indeed, brute force methods of AI/ML, computational biology, etc., will be applied to cancer cells to shotgun their entire proteosome, genome, etc., at scale. However, in the rogue application of technology to unravel the most intimate of cancer’s secrets, there will still be room for refinement; a place for elegant creativity in a world of crude thought. Accordingly, the art of drug development will play just as important a role as the science. The strategic control point will shift away from finding the right drug for a protein target to identification of the protein target itself. It will move away from conceptualizing “the right” single agent therapies to determining what “the correct” combination therapy is. Entire pharmaceutical companies will succeed or fail based on whether or not they choose the right drugs to combine with theirs, and the right tumors to test them in. The jobs of tremendously talented individuals will hinge on these decisions. Therefore, everyone in the pharmaceutical value chain, from the researcher in the lab to the CEO of the company, must focus on how to identify optimal drug targets and combination therapies. It’s my opinion the performance of pharmaceutical executives should be assessed largely by how well they choose these protein targets. In “The Insider’s Guide to Translational Medicine” we have literally provided those paying attention with the answers to some of the questions on the test [1]. We revealed the oncologist’s short-term [2] and long-term approach to cancer care [3]. We spoke of assessing the cell globally through differential multiomics [4], and did a deep dive on assessing AI/ML based claims [5]. We comprehensively revealed the profound power of cell free DNA (cfDNA) [6] and its utility in cancer [7]. Today we will bring everything together as we speak directly to the drug developer and demonstrate how cfDNA will have a central role in identifying protein targets and assessing the efficacy of drugs targeting them.
In the first two chapters of a “Hitchhiker’s Guide to Cell-Free DNA” you served as CEO of Comprehensive Precision [6, 7]. Your board asked you to present your plans for the cfDNA facet of the company in a three-day presentation. On day 1 you arduously delivered a primer to your board that garnered a standing ovation that caused you to blush [6]. On day 2 you spoke directly about population health cancer screening using cfDNA [7]. You admonished the board to reconsider their desire to enter this market due to the ferocity of the players in it. They repudiated you, prompting you to develop a rudimentary strategic plan to enter the market. Today you’re excited to dazzle the board with how cfDNA can unlock drug development secrets that can refine our therapeutic approach to cancer patients.
The Backdrop
Watching the dolphins in the distance from your “Cliffs at Princeville” condo Air BnB in Kauai (I forget to tell you this was all happening in Kauai) you wish you could be swimming among them (figure 1).

Figure 1: The “Cliffs at Princeville” in Kauai
You’re excited about the dolphin tour you have scheduled the next morning, but are keenly aware your job in Kauai isn’t complete. You were congratulated by several board members both days you left the meeting, but you’re aware we live in a Janet Jackson “What have you done for me lately?” sort of a world. Everything you accomplished the last two days could vanish with a single misstep. You’re not nervous, but you know what’s at stake.
The Uber drops you off at the Grand Hyatt where the board members are staying and the meeting is being held. You are the first to arrive at the meeting and get set up (figure 2).

Figure 2: Meeting room at the Grand Hyatt in Kauai
After exchanging pleasantries with the board members, you’re eager to show them what you prepared.
The Intro
You take a sip of your glass of water and say:
I always think of the Jeff Goldblum quote in Jurassic Park where he says "life finds a way" when I think of cancer. However, I simply substitute the word life with cancer in that "cancer finds a way". Indeed, the bane of an oncologist's existence is the heterogeneity of tumors and the numerous resistance mechanisms cancer cells employ. One of the most fundamental questions in Oncology is why we can't cure the vast majority of stage 4 tumors even though we can often eradicate large swaths of tumor cells.
The bottom line is nothing in a cell happens in a vacuum. If you perturb the MAP kinase pathway in some way there are numerous changes happening across the entire cancer cell. Accordingly, one needs to understand what is happening in the cell globally. Combination therapy is essential and rooted in anticipating these macroscopic alterations.
The misnomer here is that what we're presently doing is incredibly crude. We basically are taking whatever we have on the shelf and combining it with whatever else we have on the shelf. Right now, it's the best we can do, but we need more refined approaches.
It doesn't take a genius to say, "let's see what happens when we take olaparib and mix it with keytruda or abiraterone or whatever in prostate cancer." Anyone can add a VEGFR inhibitor to ipilimumab + nivolumab in renal cell cancer. Indeed, one can conceive of numerous permutations of clinical trials where we simply test every combination of therapies we have available. The key is to understand what is happening at the cellular level to optimize our therapeutic approach. It will also be important to determine what our efficacy measures really are. Progression-free survival without showing improved overall survival is likely insufficient as that doesn't prove that combination therapies are better than using them sequentially.
A term I coined is the "cellular equalizer". When one turns up the gain in the MAP kinase pathway what's happening to the gain in the JAK/STAT pathway, PD1/PDL1 pathway, etc. This will be incredibly important as one strives to be a conductor of another term I coined, "the cellular symphony". Indeed, this may actually allow someone to manipulate what is happening in the cancer cell to optimize cell kill.
Ultimately, when we look for resistance mechanisms we often look for overt genetic mutations, but a lot of cancer cell resistance is likely happening in a far more subtle fashion. It likely resides in the manner in which the "cellular equalizer" is modified to allow a cancer cell to survive. The issue here is how one actually measures this. The answer resides at the interface of proteomics and its various iterations, genomics, artificial intelligence, machine learning, etc. The primary obstacle in these studies will be the sheer amount of data required and the cost associated with generating the data.
In my opinion, this is the key to optimizing cancer regimens via combined therapeutics. Today we’re going to talk about cfDNA in this equation, but multiomics will play a central role.
cfDNA in the identification of drug targets and optimal combination therapies
After your intro the board members are riveted. You have them, but now you go in for the kill. You, along with very talented individuals, presented the second episode of the Revolution Cancer International Molecular Tumor Board to the world on July 30, 2022 [8]. During that talk you presented the PMV Pharma pipeline, largely comprised of PC14586, a p53 Y220C activator that restores wild-type p53 function in the mutated protein [9]. You presented an unprecedented approach to cancer treatment cartography; the practice of creating optimal patient specific treatment maps incorporating conventional therapy, surgical options, molecular based drugs, locoregional treatments, and clinical trials [3]. You applied this approach to a patient with extensive stage small cell lung cancer with a p53 Y220C mutation, and to NINE patients with stage 4 hepatocellular carcinoma (HCC) [8].
You show the molecular profile for the nine liver patients (figure 3) to the board and implore them that these 9 patients are telling the same story, even in death. All of them have amplification of 11q13.3, which houses CCND1, FGF19, FGF3, and FGF4 (figure 4). They all have the same human telomerase reverse transcriptase (hTERT) mutation; a protein you are incredibly familiar with because your Ph.D. thesis was titled “Transcriptional Regulation of Human Telomerase Reverse Transcriptase (hTERT)”. All 9 of the patients have a mutation in p53, with 8 of them having a mutation at R249 (4 R249M, 4 R249S). You note there was NTRK1 amplification in 3 of 9 patients.

Figure 3: The molecular profiles of nine patients with stage 4 hepatocellular cancer
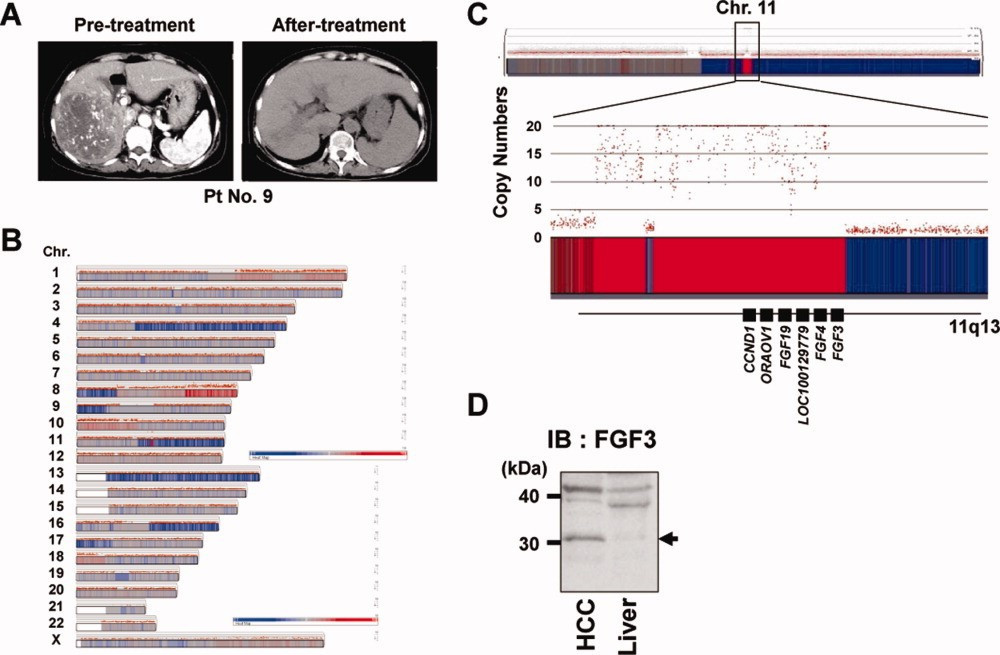
Figure 4: 11q13 containing CCND1, FGF19, FGF4, FGF3 [10]
As your presenting the nine HCC patients you can tell the board members are a bit stunned, but now you truly go in for the kill.
You reference an incredibly popular review paper detailing the “hallmarks of cancer” published by Hanahan and Weinberg in Cell in 2011 (figure 5).
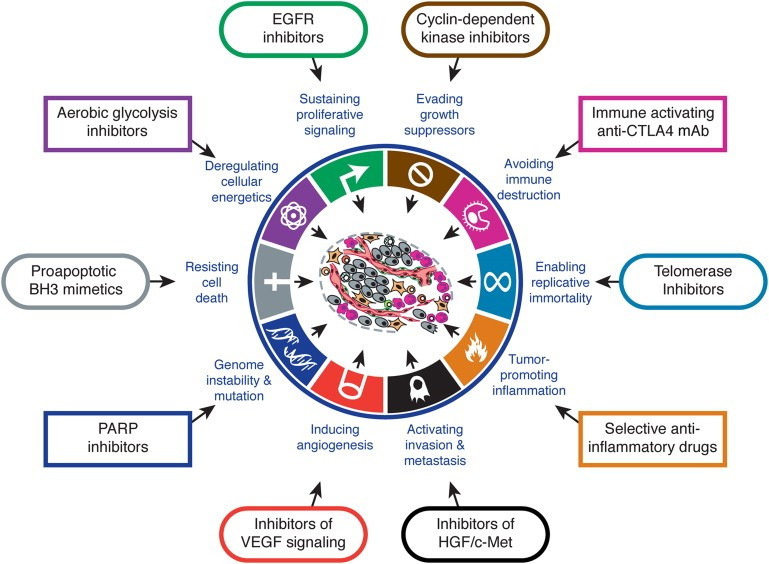
Figure 5: Hallmarks of Cancer [11]
You tell the board these will likely continue to be refined and they were already different than what Hanahan and Weinberg published years before. You speak about the cellular equalizer you discussed in your intro, and that it is a direct manifestation of the hallmarks of cancer. The hallmarks of cancer are conceptual, whereas, the cellular equalizer is tangible and constitutes all the molecular changes cancer cells undergo to proliferate in an uncontrolled manner. You illustrate that the hTERT mutation facilitates cancer cell replicative immortality. The cyclinD1 amplification promotes evasion of growth suppressors. The p53 mutation promotes genome instability and mutation, evasion of growth suppressors, resistance to cell death, etc. FGF19 amplification promotes invasion and metastases, and sustains proliferative signaling. As such, you tell your board the cancer cell is likely too smart to be defeated by single agent therapy. Even where single drugs are remarkably effective, such as cancers with NTRK fusions, BCR-ABL mutations, EGFR exon 19 or exon 21 L858R mutations, ALK fusions, ROS1 fusions, BRAF V600E mutations, etc., the corresponding drugs do not “cure” patients. Indeed, you strongly feel companies that can identify the “right combination therapies” will be extremely successful in the future. Sadly, you know that's easier said than done.
You have often wondered if we abandon drugs we shouldn’t be because they weren’t tested in the right trial. You question the logic of therapies being required to demonstrate efficacy as a single agent before they can be trialed in combination. However, you do note that some recent trials suggest this paradigm may be changing. Astra Zeneca has a trial in stage 3 non-small cell lung cancer looking at durvalumab alone, durvalumab plus CD73 AB, durvalumab plus CD94 AB, and durvalumab plus CD73 AB and CD94 AB. You’re curious how the FDA approved the trial as CD73 and CD94 haven’t been shown to be effective as single agents, but you hope this is the beginning of a trend.
You are keenly aware there is big push to salvage drugs from the trash heap of oncology trials. Many are going back to these drugs to repurpose them for use in other medical disciplines.
Based on the aforementioned molecular profiles, you ask your board, “what if we could target cyclin D1 amplification, the p53 variant, and hTERT at the same time?” Indeed, it is your firm belief the future resides in ultra-personalized therapies that will unlock the true value of precision medicine. Memorial Sloan Kettering is heavily involved in the production of bespoke CAR-T cellular therapies. Why not do the same with drug trials? Of course, you’re aware the costs of clinical trials prohibit companies from developing drug cocktails specific to each patient, but you hope this changes in the future. Nonetheless, you soldier on.
To send your message home you elect to demonstrate in real-time how you would use the aforementioned cfDNA HCC data to devise ENTIRELY NOVEL clinical trials. You tell the board based on the data one could propose refined HCC clinical trials, including:
- Tecentriq+Avastin+/-CDK4/6 inhibitor in first-line treatment of metastatic HCC patients with 11q13.3 amplicon (with concurrent liquid biopsy) AND proficient RB1 expression.
Tecentriq+avastin is a commonly used regimen for the first-line treatment of stage 4 hepatocellular cancer that are classified as Child-Pugh A in severity. It is very well tolerated, but has a median progression free survival of approximately 7 months and an overall response rate of 27% [13]. Unfortunately, it’s the best we have in first-line HCC, but what if we can do better? You postulate that adding a CDK4/6 inhibitor, such as palbociclib, ribociclib, or abemaciclib, could be extremely beneficial in HCC patients that have the cyclin D1 amplicon, which is approximately 5-7% of stage 4 HCC patients [10]. Importantly, you note these patients would need to have intact RB1 function, which is mutated in 30% of HCC patients [12], to be affected by a CDK4/6 inhibitor. - Ipilimumab+Nivolumab or Durvalumab+Tremelimumab+/-CDK 4/6 inhibitor in first-line treatment of metastatic HCC patients with 11q13.3 (with concurrent liquid biopsy) AND proficient RB1 expression.
Ipilimumab+nivolumab and durvalumab+tremelimumab are immunotherapy regimens involving CTLA4 (ipilimumab and tremelimumab) and PD-1/PD-L1 inhibitors (nivolumab and durvalumab). They are designed to stimulate the immune system to eradicate cancer cells. Ipilimumab+nivolumab is approved in the second-line treatment of stage 4 HCC patients and has an overall response rate of 32% [14]. Durvalumab+tremelimumab will likely be approved in the first-line setting with an overall response rate of 20.1%, median overall survival of 16.56 months and 3-year overall survival rate of 31% [15]. To this end, it’s certainly nice to have numerous options for our stage 4 HCC patients, but we clearly need to do better. One could consider a trial with either of these regimens +/- a CDK 4/6 inhibitor with stratification based on the cyclin D1 amplicon and RB1 expression. - Phase 1 UV1 (hTERT vaccine)+tecentriq+avastin+CDK 4/6inhibitor+p53 inducer (with concurrent liquid biopsy)
No two cancer patients are the same. They have different tumor burden, stages, locations of metastases, histology, cancer biology, cancer cell molecular profiles, protein expression profiles, etc. Yet, the vast majority of clinical trials treat cancer patients as though they are homogeneous. This is understandable as clinical trials cost a fortune to conduct. However, we must find a way to personalize our treatments. This mandates we find a way to do personalized clinical trials. The precision medicine clinical trials, such as MATCH, TAPUR, etc., are a nice start, but are NOWHERE NEAR where we need to be.
Consider the nine patients presented here. It would be ideal to do a study that combines an hTERT inhibitor, p53 activator, and CDK 4/6 inhibitor superimposed on a tecentriq/avastin backbone if possible. Indeed, the data presented here suggests that one should consider developing a p53 R249S inducer, as p53 R249S mutations are often seen in patients with HBV related HCC. - Lenvatinib+pembrolizumab+/-CDK4/6 inhibitor (with concurrent liquid biopsy) in patients with 11q13.3 amplicon.
Lenvatininb is approved in the first-line treatment of stage 4 HCC with a 24.1% overall response rate, median overall survival of 13.6 months, and a median progression free survival of 7.3 months [16]. It is a VEGFR inhibitor, but has numerous off target effects, including inhibition of FGFR1, 2, 3, and 4. FGF19, which is amplified in every single patient presented here, activates FGFR2. Thus, it would be nice to conduct a trial using lenvatinib and a CDK 4/6 inhibitor in the aforementioned patients. In addition, since we know lenvatinib and pembrolizumab can be safely given together in stage 4 renal cell cancer and endometrial cancer, a trial exploring lenvatinib plus pembrolizumab +/- a CDK 4/6 inhibitor may prove useful in patients with the 11q13.3 amplicon and intact RB1 expression.
After presenting your clinical trial ideas to the board you can see their eyes glazed over. You very quickly tell them the details of what you just said DO NOT MATTER. You were simply conducting a real-time exercise to illustrate the POWER of cell-free DNA in drug development and clinical trial design.
From a drug development perspective, the nine patients you presented suggest that a p53 drug development program targeting the R249S variant may be warranted, as opposed to targeting other p53 variants. If one could develop a drug that simultaneously restored p53 wild-type activity in p53 R249S and R249M variants, seen in 8 of the 9 HCC patients presented, it could be incredibly beneficial and lucrative. Moreover, the patients discussed reveal the power of cfDNA in designing a refined clinical trial design and optimal combination therapy, although it may ultimately still prove ineffectual.
You tell the board that what you showed them was the tiniest microcosm of the power of cfDNA in drug development. You ask them to envision a scenario where you can take hundreds of thousands of patients with HCC and look at the cancer cell protein, RNA, lipid, DNA, and carbohydrate profile. Today you’re focusing on cfDNA, but you tell your board to envision what you just discussed at an exponential level.
You emphasize to the board people are already doing what you just described. You speak about Illumina’s recent entry into drug development. You talk about Tempus and how they’re partnering with pharmaceutical companies. And for the coup de gras, you tell them about a slew of companies that received millions of dollars to develop entire drug target identification platforms based on the very simple methodologies you described. Indeed, you tell them there is a minimal barrier to entry in this market, and it’s rapidly becoming saturated. Just like you told them the last two days when talking about population health cancer screening, the primary strategic control point in this arena will be who has the tissue and the data. UPMC is well aware of this and speaks directly to it being one of their strategic advantages [17]. Dana Farber, MSK, MD Andreson, etc., have leveraged their patients’ data in astronomical ways. Vanderbilt basically just sold the use of their biobank to Illumina [18]. And the list goes on and on…
Ultimately, you tell the board that all you did with the nine liver cancer patients you presented was pattern recognition. You tell them that AI/ML and having enough patient samples, lets you conduct “pattern recognition on steroids”. And as facile as it is from a scientific and data science perspective, it’s an incredibly powerful and lucrative tool. It’s why essentially EVERY biotech company is rapidly building robust data science programs. It's why you feel EVERY single person in biotech should become familiar with AI/ML, and you recommend the MIT Great Learning Applied Data Science Bootcamp (figure 6) you took previously if they don’t want to formally complete a Master’s degree in the discipline [19].
Figure 6: MIT/Great Learning Applied Data Science Bootcamp [19]
You tell the board that where we are now will be unrecognizable in the future. Google just released the structure of 200 million proteins… 200 MILLION [20]. Nvidia is fully entrenched in the drug development space, and many companies have AI/ML based drug design programs. Accordingly, the key will be in identifying the right drug target and combination therapies. cfDNA is a powerful tool in this regard, and you feel strongly that drug developers would do well to recognize this if they haven’t already. Indeed, they are already late to the party.
cfDNA in assessing drug efficacy in clinical trials
After delivering the first half of your talk, you watch as the board members digest it. You sense they are a bit overwhelmed, despite being smart and talented individuals. You walk to the center of the desk, grab the water pitcher, and poor yourself a glass. After taking a few sips you’re ready to get back to it.
When you press the mouse your next slide titled, “Taking multiple shots on goal through cfDNA” appears. And with that you launch into a diatribe regarding the importance of running companion biomarker studies in any drug clinical trial.
It’s conceivable the drug you develop only works on cancers with a certain genetic composition. Failure to run companion genomics while testing your drug in clinical trial can be catastrophic. It can mislead you into believing the drug is a failure, whereas it may have been a resounding success in patients that harbor tumors with specific molecular aberrations. Consider the recent examples of alpelisib, elacestrant, and PC14586.
Alpelisib is a PIK3CA inhibitor used to treat the 40% of stage 4 hormone receptor positive, HER2 negative or low-positive, breast cancer that harbor PIK3CA mutations. In the seminal study of alpelisib in stage 4 breast cancer published in the NEJM in 2019, median progression free survival (PFS) was 11 month for alpelisib-fulvestrant versus placebo-fulvestrant in patients with PIK3CA mutated tumors [21]. In patients without PIK3CA mutation median PFS was 11 months for alpelisib-fulvestrant versus 5.7 months for placebo-fulvestrant. Accordingly, the FDA approved alpelisib-fulvestrant for second-line treatment of PIK3CA mutated, stage 4 hormone receptor positive and HER2 negative breast cancer in 2019
Elacestrant, like already FDA approved fulvestrant (faslodex), is another selective estrogen receptor degrader (SERD) that was recently submitted to the FDA for approval of third-line stage 4, hormone receptor positive, HER2-negative breast cancer [22]. The median progression free survival with elacestrant in the supporting study was 2.79 months versus 1.91 months for standard of care comprised of endocrine therapy. Based on this data alone I don’t feel elacestrant has a shot of being approved because the PFS is minimally improved and the standard of care arm used was endocrine therapy, which would rarely be relied on in this setting. However, Radius Health who makes the drug, was smart. They stratified their data based on whether or not a patient’s tumor had an ESR1 mutation. ESR1 mutations typically confer resistance to endocrine therapies targeting the estrogen receptor, and were observed in 48% of patients in the Radius Health study. Fortunately for Radius Health, median PFS was 5.3 months for elacestrant versus 1.9 months for ”standard of care” endocrine therapy in the ESR1- mutation positive breast cancer patients. Accordingly, whereas elacestrant likely would have been dead in the water without the companion ESR1 mutation testing, it has a chance of being approved by the FDA for ESR1 mutated, hormone-receptor positive, HER2 negative/low-positive stage 4 breast cancer.
PMV Pharma developed PC14586 to restore P53 wild-type function in patients with p53 Y220C mutated cancers [9]. The drug works by intercalating into the crevice in p53 created by the Y220C missense mutation (figure 7).
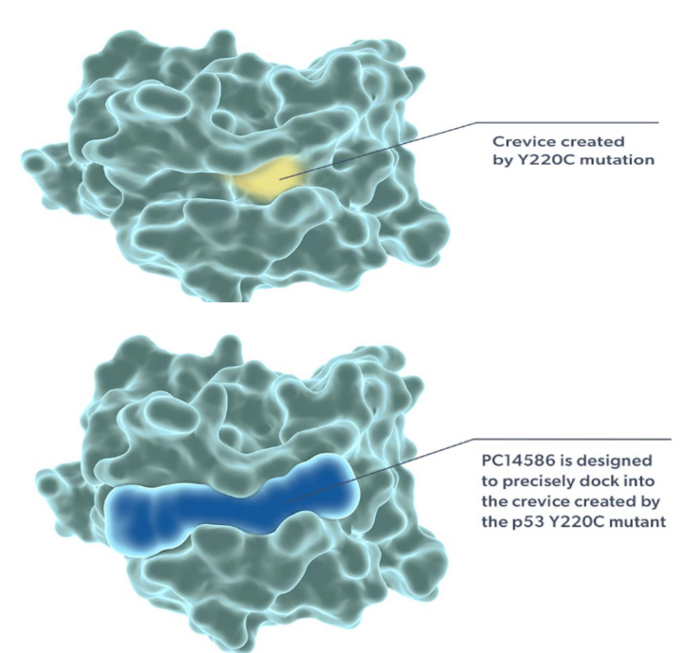
Figure 7: PC14586 docks in the crevice created by the p53 Y220C mutant
When cancer cells harboring p53 Y220C mutations are treated with PC14586, wild-type p53 function is restored as MDM2 and p21 expression is increased (figure 8). In addition, nude mice implanted with p53 Y220C mutated cancers have regression of their tumors when treated with high concentrations of PC14586, presumably due to restoration of wild-type p53 function.
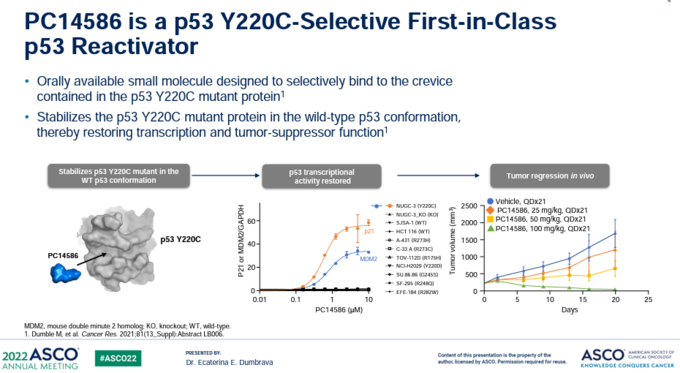
Figure 8: PC14586 restores wild-type function to p53 Y220C by filling the crevice created by the mutation (left), resulting in increased MDM2 and p21 expression in p53 Y220C mutated cancer cells, and reduced tumor growth in mice implanted with p53 Y220C mutated cancer cells.
In a phase 1/2 trial of PC14586 in patients with advanced solid tumors harboring a p53 Y220 mutation PC14586 had an overall response rate of 32% at the higher doses (figure 9) [9].
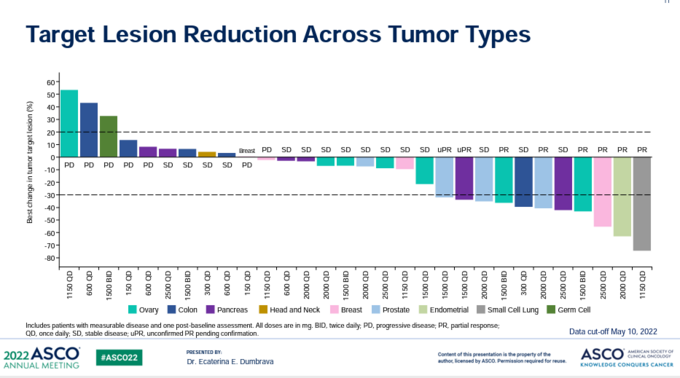
Figure 9: Waterfall plot of patients with p53 Y220C mutated tumors treated with PC14586.
Many patients on trial had progression free intervals of over 4-5 months and remain on the drug. Importantly, PMV Pharma could track the effect of their drug on p53 Y220C mutated proteins by assessing circulating tumor cells and cell tumor DNA in the blood (figure 10). They showed both of these decreased when patients were treated with PC14586, presumably due to cancer cells dying.
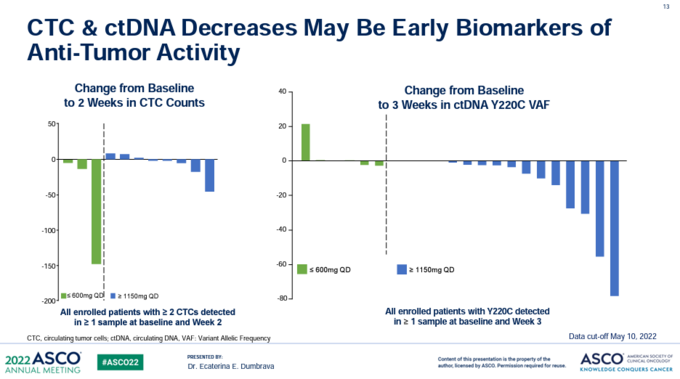
Figure 10: p53 Y220C mutated circulating tumor cells and cfDNA decrease when patients are treated with PC14586.
Collectively, the aforementioned three examples constitute the “tip of the iceberg” when considering the power of genomics, and cfDNA specifically, in testing the efficacy of new therapeutics. Companion genomics give companies multiple shots on goal as it enables them to look for patient subsets that respond favorably to their drug even if the trial was globally negative. Failure to run companion genomics precludes companies from molecularly profiling tumors and correlating this with patient outcomes. This could prove catastrophic, which is why I feel it’s imperative every drug clinical trial must have a genomics component and consider multiomics based biomarker assessments. Ideally, patient samples would be obtained at numerous intervals during the trial to frequently assess the cancer’s molecular profile. Doing this may enable a company to identify genomic treatment and resistance signatures that could be invaluable, and lead to drug approval.
An End to the Three Day Saga
After showing the board how powerful cell free DNA is in drug development you harken back to the first day you spoke to them. You remind them that cfDNA has numerous potential applications in cancer (figure 11), including:
- Population Cancer Screening and Early Intervention
- Residual Disease Measurement
- Cancer Surveillance
- Predict Treatment Response
- Prognostication
- Account for Tumor Heterogeneity
- Dictate Treatment
- Identify New Drug Targets
- Drug Development
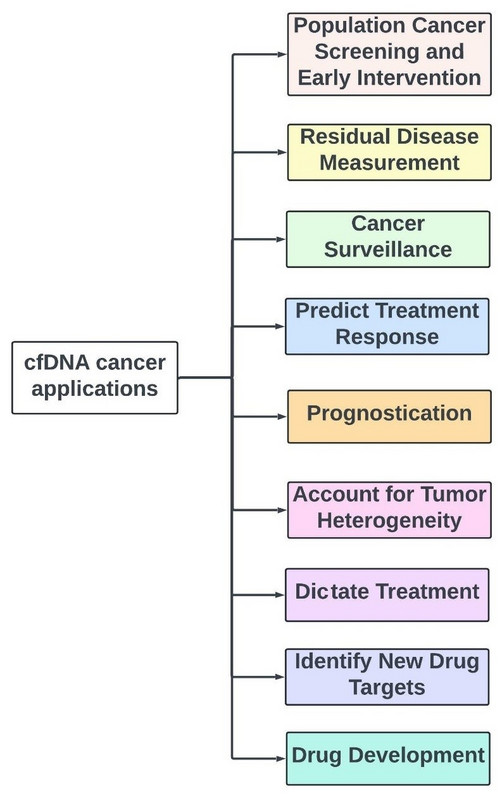
Figure 11: Utility of cfDNA in cancer care.
You quickly recap the talk you gave the day before on cfDNA in Population Health Screening and Early Intervention [7], and emphasize that someday we will be getting annual cancer screening cfDNA tests. You tell them cfDNA already pervades numerous arenas in medicine, but will become truly ubiquitous in the immediate future. Accordingly, ignoring cfDNA is untenable if you’re involved in healthcare in any capacity. You refer them back to the primer you presented on day 1 of the meeting, and thank them for their time and attention [6].
You ask if any of the board members have any questions, and are relieved when they don’t. Purely on accident, the mic slips out of your hand, but you feel like it’s an appropriate metaphor for the truth you laid down the last three days. You pick up the mic, put it on the table, and thank the board for their time.
As you leave the meeting room and walk through the Grand Hyatt lobby to the exit you can finally exhale. You don’t get nervous, but you’re relieved to be done. You get in the Cadillac Escalade Uber waiting in front of the hotel and head for your condo rental. You run into the condo, change your clothes, and get back in the Cadillac Escalade. The driver drops you off at the beach where you rent a surfboard. As you paddle out to catch a wave and think about the dolphin excursion you have planned for the morning, you gaze out across the ocean and nothing else matters…
Conclusion
This marks the end of the three chapter “Hitchhiker’s Guide to Cell-Free DNA”. Honestly, this mini-series got away from me. I didn’t expect to go down the cfDNA rabbit hole, but when I did, I couldn’t get out. Indeed, cfDNA is becoming so ubiquitous a three chapter expose on the subject hardly does it justice. It’s imperative the reader be cognizant of how powerful cfDNA truly is, particularly given its relative simplicity.
As people we’re a conglomeration of our experiences. We are what we’ve lived. To this end, having been a lot of places, and having an MD, Ph.D., MBA, and formal training in AI/ML, has afforded me the opportunity to experience numerous worlds. I’ve spent years of my life dreaming about experiments I would do the next day in the lab. I’ve seen months of my life disappear at the hands of a negative result. I’ve lost years of work after being “scooped” on more than one occasion. I was awarded several grants, but rejected for several others. I know what it’s like to be a basic science researcher and a clinician.
I know basic scientists generally don’t spend a lot of time considering the clinic and many clinicians don’t consider the science behind what they do. I find it ridiculous that oncologists become famous and rich for simply giving patients drugs, while the individuals that developed the drugs go frequently unrecognized.
The world isn’t fair. We don’t live in a meritocracy. All too often how you say something matters more than what you say. Who you know matters more than what you know. Titles matter even when they are largely meaningless and shouldn’t. Talented individuals who should be recognized for their contributions to patients forever go unnoticed. It’s sad, it really is. And I actually think it will get worse for the drug developer in the future.
As AI/ML makes drug development more accessible to people with minimal related training, drug developers need to reinvent themselves. They need to recognize one of the primary strategic control points in the future will be identifying the right proteins to target, right tumors to test them in, and the right combination therapies to employ. A basic understanding of what’s transpiring in the clinic can help drug developers see the forest and successfully ideate.
This is the seventh entry in “The Insider’s Guide to Translational Medicine”. I have tried my best to give the reader the answer to some of the questions on the test, so to speak. We have traveled at light speed from the clinic to data science to cell free DNA, all while remaining forever cognizant of the forest and not getting lost in the trees. I sincerely hope you’ve enjoyed the journey so far. However, I encourage you to buckle up. Your work as CEO of Comprehensive Precision is nowhere near done…
References
1. https://www.biopharmatrend.com/topic/the-insiders-guide-to-translational-medicine/posts/.
2. https://www.biopharmatrend.com/post/510-quarterbacking-a-patients-cancer-care-for-submission/.
5. https://www.biopharmatrend.com/post/523-remember-to-be-precise/
7. https://www.biopharmatrend.com/post/561-hunting-cancer-before-it-can-hunt-you/
8. https://www.youtube.com/watch?v=zQL93TxDIFU&t=134s
9. pmvpharma.com
10. Arao T, et. al, Hepatology. 2013 Apr;57(4):1407-15
11. D Hanahan and RA Weinberg, Cell, Vol 144, March 4, 2011
12. Niu et al, Journal of Cancer, 2019
13. Finn et al, NEJM, May 14, 2020
14. Yau T, Kang Y, Kim T, et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020;6(11):e204564. doi:10.1001/jamaoncol.2020.4564
15. Chassan et al, NEJM, June 6, 2022.
16. Kudo et al, Lancet, March 24, 2018
17. https://endpts.com/exclusive-upmc-launches-another-biotech-rooted-in-the-tumor-microenvironment/
19. https://www.mygreatlearning.com/mit-data-science-program
21. Andre et al, Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. NEJM. 2019; 380: 1929-1940.


