Predicting a Patient’s Future with a Crystal Ball Comprised of Cell-free DNA
Chapter 1 of a 3-chapter Hitchhiker’s guide to cell-free DNA for travelers of all backgrounds and expertise.
Even though I’ve been a hematologist/oncologist for over a decade, I’m horrendous at dealing with death. It still shatters me every time I have to tell a patient they’re going to die of their cancer. It’s supposed to. If it didn’t, I need to find another job.
There have been clinic days where I’ve seen 30 patients, 17 of which had theoretically incurable cancers. Accordingly, I’m on record as saying nobody would be happier than me if my skill set was no longer needed by society.
It’s often forgotten that NO TWO CANCER PATIENTS ARE THE SAME. They have different tumor types with different histology, molecular profiles, stages, comorbidities, etc. Accordingly, in a world so heterogeneous, there are next to no generalities in cancer. However, there is a singular universality, “the earlier the better”. The sooner I know about a patient’s fever, infectious symptoms, etc., the more likely they will do well. The earlier we identify a patient’s cancer the more likely they can be cured.
In this editorial series we’ve moved at breakneck speed as we talked about the long- and short-term approach to cancer care, and how to assess the claims of people in healthtech, genomics, pharmaceuticals, science, etc. [1, 2, 3]. Today we begin to put the entire cell-free DNA cancer screening industry under the microscope. We will assess the grandiose claims of numerous companies that they are the solution and will enable us to get one step closer to putting me out of work. In so doing, we will draw on the lessons conveyed in the prior “Drowning in Data”, “Quarterbacking a Patient’s Cancer Care”, and “Playing Chess Against Cancer” entries in this editorial series, as we take a breathtaking tour of the cell-free DNA world with you as CEO.

Image credit: Adrienn Harto
It Begins...
It’s well established that cell-free DNA (cfDNA) holds significant promise in assessing mutations in the fetus, identifying and surveilling cancer, predicting a patient’s response to treatments, identifying early signs of solid organ transplant rejection, etc. Yet, this is just the tip of the iceberg as cfDNA will pervade every facet of the medical world. Accordingly, it’s imperative everyone involved in healthcare, including healthtech, pharmaceutical, biotechnology, and clinic employees, have some familiarity with cfDNA.
A discussion of all potential medical applications of cfDNA would be endless. An exhaustive writing of the putative utility of cfDNA would be several volumes long. Accordingly, we will primarily focus on the use of cfDNA in hematology and medical oncology, largely because it’s my areas of expertise.
Our tour of cfDNA in blood and cancer diseases will require no previous understanding of molecular biology, and is geared towards everyone. For the reader who has no medical background, we begin our discussion reviewing the basics of DNA, transcription, chromatin, etc., en route to explaining how cfDNA tests are applied in cancer. For the cfDNA specialists, I will ultimately provide a list of NEARLY EVERY SINGLE TUMOR TYPE, and where I personally feel the greatest utility of cfDNA could be. I will detail potential clinical trials in this regard. For the investor and new entrants to the cfDNA space, I will provide a comparative analysis of companies in the cfDNA cancer market, and perceived strategic control points in the field. Indeed, I recently advised two cfDNA start-ups in this regard; I will not be revealing any of their secrets.
Ultimately, our tour of cfDNA will be three chapters long, this being the first:
- Chapter 1: An introduction to cfDNA and overview of cancer related applications.
- Chapter 2: cfDNA in population cancer screening.
- Chapter 3: cfDNA in cancer prognostication, surveillance, and treatment.
CEO of Comprehensive Precision
Throughout the remainder of the “Insider’s Guide to Translational Medicine” series you will serve as CEO of a company called Comprehensive Precision. As the name implies, your company specializes in multiomics, including proteomics, genomics, lipidomics, RNAomics, etc., and has multiple branches (figure 1).
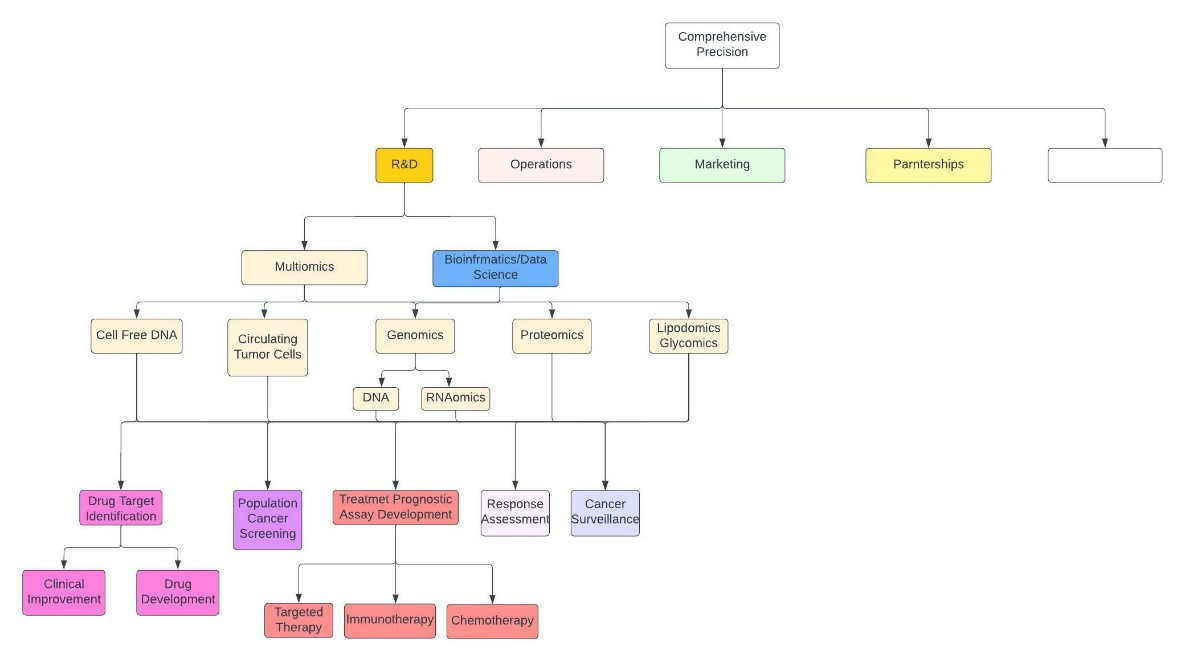
Figure 1: Abbreviated and crude description of Comprehensive Precision. The empty white box encompasses everything else.
Today you’re been summoned to a board meeting to discuss your strategy for the cell-free DNA facet of your corporation (yellow box in figure 1).
Paging Elizabeth Holmes?
The claims being made by cfDNA companies, namely GRAIL, seem too good to be true. They are eerily reminiscent of Elizabeth Holmes in peak Theranos times. Indeed, GRAIL claims they can “sufficiently” determine if you have one of 50 cancers from a single blood sample. Yet, they are not alone. Exact Science, Freenome, Delfi, etc., have made substantial claims of being able to detect various cancers long before they become clinically apparent.
As you prepare to present to your board you note some of the members are not scientifically inclined. Therefore, before speaking about Comprehensive Precision’s cfDNA cancer-related plans, you provide them with Chapter One of our “Hitchhiker’s guide to cell-free DNA”.
Chapter One: DNA
Deoxyribonucleic acid, DNA, is perhaps the single most important macromolecule on the planet. It is comprised of two polynucleotide chains that coil around each other to form a double helix (figure 2).
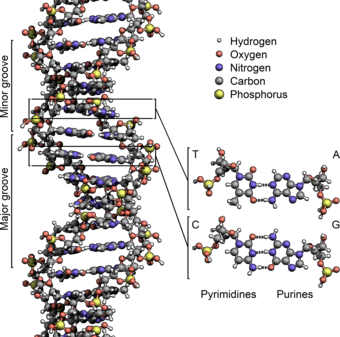
Figure 2: Representation of DNA illustrating A, T, G, and C nucleotides (4).
It carries genetic instructions for the development, functioning, growth, reproduction, etc., of all known organisms and many viruses (4). Each DNA polynucleotide is comprised of nucleotides that consist of a nucleobase, a sugar called deoxyribose, and a phosphate group. There are four type of nucleotides, cytosine (C), thymine (T), guanine (G), and adenine (A), that are named after their corresponding nucleobases (figure 3). Thymine and Cytosine belong to the pyrimidine group, and the purine group includes adenine and guanine (5).
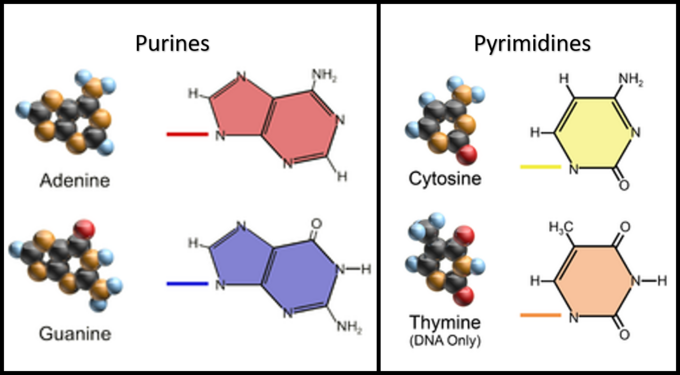
Figure 3: Structure of DNA nucleobases (5).
Nucleotides in each polynucleotide chain are joined to one another by phosphodiesterase bonds, that covalently bind the phosphate group of one nucleotide to deoxyribose of the adjacent one (6). This forms an alternating phosphate-deoxyribose DNA backbone (figure 4).
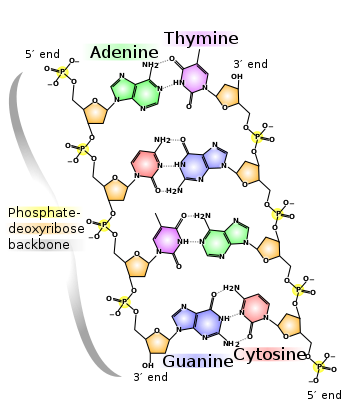
Figure 4: Structure of DNA with special attention to the phosphodiesterase bond (5).
The two separate polynucleotide strands of DNA are bound together via hydrogen bonds between the nucleobases of each nucleotide (figure 4). This association follows base pairing rules in which A binds to T and C with G.
DNA to mRNA to Protein
Just like blueprints that outline the construction of a building, DNA is primarily a set of instructions that manifests as RNA and protein. It contains numerous genes, which are sequences of nucleotides encoding the synthesis of RNA (figure 5).
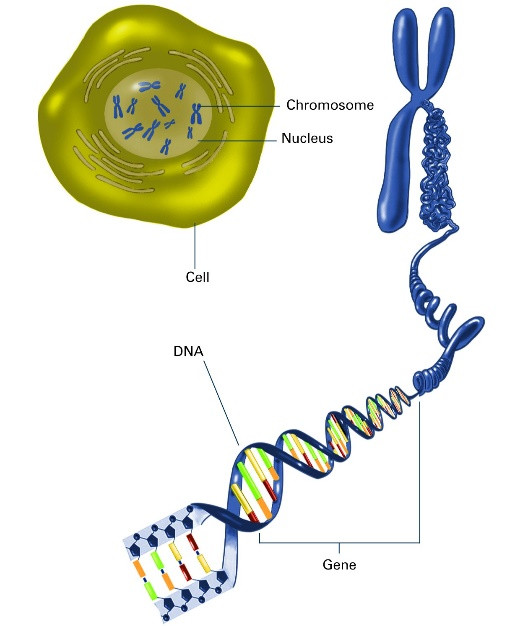
Figure 5: Illustration of a gene in the context of DNA (6).
A central tenet of life is that DNA is transcribed to messenger RNA (mRNA) in a process called transcription, and mRNA is translated into protein in a process called translation. Nowadays, we know this rubric is remarkably oversimplified as DNA codes for far more than mRNA, a topic we will explore in a later article.
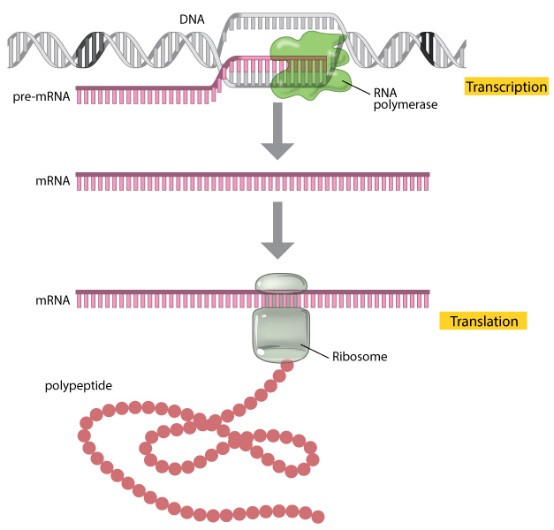
Figure 6: DNA to mRNA to protein paradigm (7).
Chromatin
DNA resides in the nucleus of cells where it is complexed with octamers comprised of proteins called histones (two each of Histone H2A, Histone H2B, Histone H3, and Histone H4), and stored in a structure called chromatin (5). This facilitates packaging of long DNA molecules into compact, denser structures, which are essential for preventing DNA damage, regulating gene expression and DNA replication, etc.
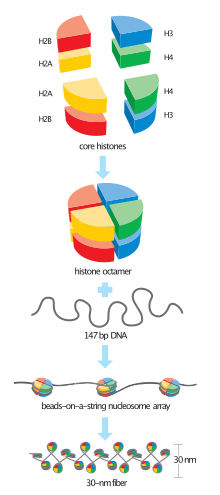
Figure 7: DNA associates with an octamer of histones to form chromatin (8).
In humans, chromatin is generated in a three-fold process. Initially, approximately 147 base pair dsDNA wraps around histone cores, comprised of eight protein subunits (two each of H2A, H2B, H3, and H4 histones), forming nucleosomes (figure 7). These are interconnected by sections of 20-60 base pair linker DNA and a linker histone H1, with a “beads on a string” appearance, and referred to as euchromatin. Multiple histones then wrap into a 30-nanometer fiber consisting of nucleosome arrays in their most compact form, referred to as heterochromatin (figure 8). Regions of DNA containing genes that are actively transcribed (transcriptionally competent) are less tightly compacted as euchromatin. Regions containing inactive genes (transcriptionally silent) are generally more condensed as heterochromatin.
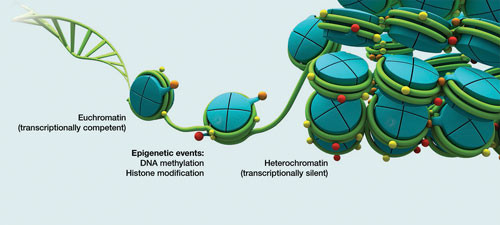
Figure 8: Euchromatin and heterochromatin (9).
The chromatin structure is highly malleable and changes during cellular process, such as cell division, DNA replication, gene transcription, etc. (figure 9). For example, during interphase of the cell cycle, in which cells replicate to form daughter progeny, chromatin is loosely structured to allow access to DNA polymerases that replicate DNA, and RNA polymerase that transcribes DNA to mRNA. DNA supercoiling of the 30-nm fibers produces metaphase chromosomes during mitosis and meiosis (figure 9).
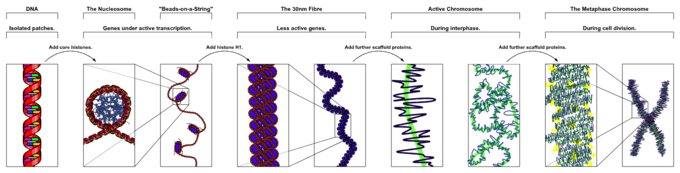 Figure 9: Chromatin changes during the cell cycle (8).
Figure 9: Chromatin changes during the cell cycle (8).
Epigenetics
Epigenetics, sometimes referred to as epigenomics, focuses on changes in DNA that don’t involve alterations to the underlying sequence (10). Two predominant epigenetic changes in humans include DNA methylation and histone acetylation (figure 10), which can regulate gene expression and alter local chromatin structure, respectively (11).
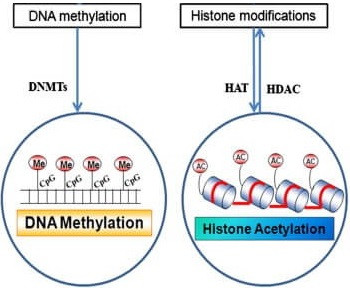
Figure 10: DNA methylation and histone acetylation impact gene expression and chromatin structure (11).
Histone Acetylation
Most modifications of chromatin occur on histone tails. In the simplest sense, histone acetylation, where acetyl groups are transferred from acetyl coenzyme A to lysine residues in the histone core of nucleosomes, results in loosening and increased accessibility of chromatin for replication and transcription (figure 11).
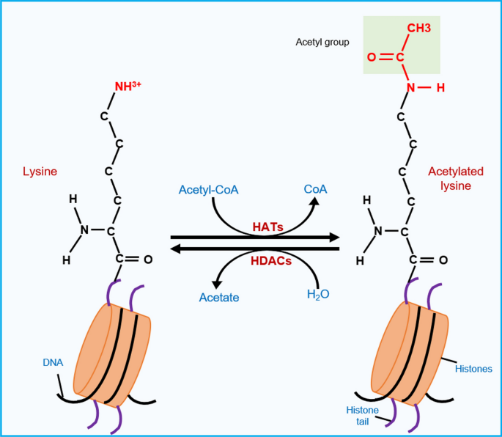
Figure 11: Histone acetylation involves addition of an acetyl group to lysine residues of histone proteins, whereas histone deacetylation is the reverse (12).
This process is catalyzed by enzymes called histone acetyltransferases (HATs). Conversely, in a process catalyzed by enzymes called histone deacetylases (HDACs), histone deacetylation results in closed chromatin and reduced gene transcription (figure 12).
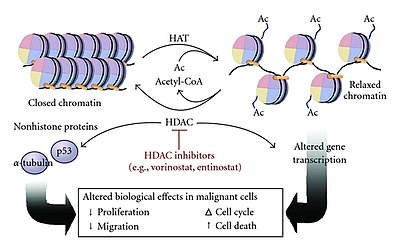
Figure 12: Histone acetylation leads to relaxation of chromatin, whereas histone deacetylation results in closed chromatin. Numerous histone deacetylase inhibitors (HDACs) are currently used, or in clinical trials, in oncology (13).
Alterations in histone acetylation equilibrium has been implicated in oncogenesis through altered gene expression, resulting in cell proliferation and migration, and reduced cell death. Many HDAC inhibitors, have been, or are presently approved, to treat various cancers, including vorinostat (cutaneous T-cell lymphoma), romidepsin (peripheral T-cell lymphoma), panobinostat (myeloma), etc. Many others, such as entinostat, are currently being tested in clinical trials.
DNA Methylation
Another type of epigenetic modification is DNA methylation, in which methyl groups are added to DNA (figure 13). DNA methylation acts to repress DNA transcription to mRNA. It plays an essential role in genomic imprinting, X-chromosome inactivation, aging, and carcinogenesis.
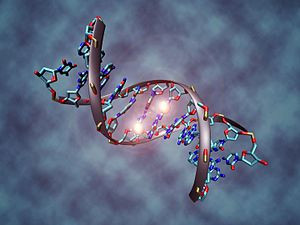
Figure 13: Representation of a DNA molecule that is methylated. Methyl groups, represented as white spheres, are shown here bound to two cytosine nucleotides (14).
In humans, DNA methylation primarily occurs on adenine and cytosine, forming N6-methyladenine, and 5-methylcytosine or N4-methlcytosine, respectively. Most commonly, it involves the addition of a methyl group to the carbon-5 position of cytosine residues, forming 5-methylcytosine (figure 14).

Figure 14: Methylation of cytosine, forming 5-mehtylcytosine (14).
In humans, methylation of cytosine, catalyzed by enzymes called DNA methyltransferases (DNMTs), occurs almost exclusively on cytosines that are contiguous with a guanine in the DNA sequence, and linked by a phosphodiester bond (figure 15). This dinucleotide region of DNA is labeled CpG, where C is cytosine, G is guanine, and p stands for the phosphodiester linkage.

Figure 15: Methylation occurs at CpG sites (left), but not at standalone cytosines (right) (15).
In mammalian somatic cells approximately 75% of CpG dinucleotides are methylated. However, CpG dinucleotides present in CpG islands (figure 16), defined as greater than 200 DNA base pairs (bps) with a G+C content greater than 50% and a ratio of observed to expected GC content greater than 0.6, are generally unmethylated (15).

Figure 16: CpG dinucleotides are in yellow. CpG islands (left) are generally unmethylated, whereas, non-CpG island CpG dinucleotides (right) are usually methylated (16).
There are approximately 25,000 CpG islands in the human genome, 75% of which are less than 850 bps. 50% of CpG islands are located in gene promoters (figure 17), which are regions of DNA usually located upstream of the genes they regulate. Proteins called transcription factors bind to gene promoters, and facilitate recruitment of other proteins, including RNA polymerase, to facilitate transcription of a corresponding gene (17).
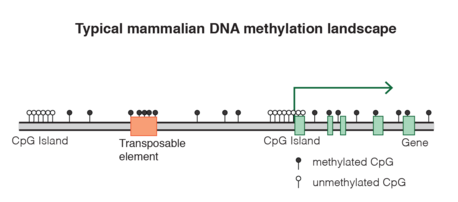
Figure 17: Promoters (green arrows) are regions of DNA usually located upstream of genes that help regulate transcription. Promoters are largely comprised of CpG islands that are generally unmethylated (white circles), whereas, CpG dinucleotides outside of CpG islands are generally methylated (black circles) (18).
The reason it’s essential promoters be heavily comprised of CpG islands is because differential methylation of these islands is a central mechanism cells utilize to regulate gene transcription. Classically, when the gene promoter is unmethylated, transcription factors, proteins that bind to gene promoters to facilitate transcription, and RNA polymerase, can access the DNA and initiate transcription. In contrast, DNA methylation can impede binding of transcription factors to gene promoters, thereby suppressing transcription (figure 18a). In addition, methylated DNA can be bound by proteins such as methyl CpG binding domains (MBDs), that impede transcription factor promoter binding, and can recruit histone deacetylases (HDACs) to generate heterochromatin to further impede transcription (figure 18b). Accordingly, methylation of promoter regions inversely correlates with gene expression. CpG dense promoters of actively transcribed genes are never methylated. With that said, our understanding of promoter methylation in transcription is more nuanced than this and evolving. Indeed, many transcriptionally silent genes have unmethylated promoters.
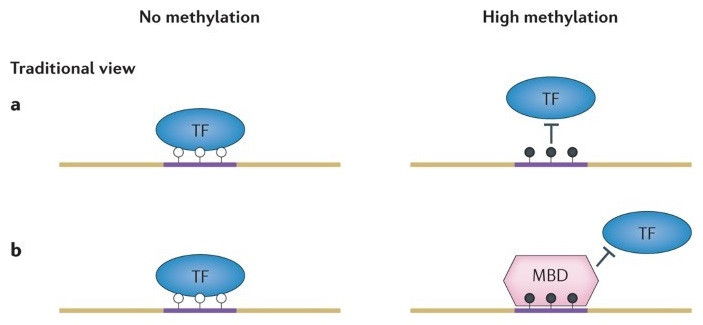
Figure 18a) Unmethylated CpG islands of promoters (white circles) facilitate transcription factor binding of promoters, whereas, methylation of promoters (black circles) impedes transcription factors from binding the promoter. 18b) Methylated CpG moieties can be bound by MBDs that further impede transcription factor binding of promoters (19).
DNA Methylation in Cancer
Cancer is just a cell that grows in an uncontrolled way. Normally, cells divide when they’re supposed to, and don’t divide when they’re not supposed to. To this end, cell division is regulated by a myriad of proteins encoded by numerous genes.
Tumor suppressor proteins, such as p53, are encoded by tumor suppressor genes, and negatively regulate cell division. Inactivating mutations in tumor suppressor genes, commonly seen in cancer, lead to reduced production of tumor suppressor proteins, resulting in uncontrolled cell division and cancer. In contrast, oncoproteins, derived from oncogenes, promote cell proliferation. Activating mutations in oncogenes, frequently implicated in malignancies, lead to unregulated cell proliferation and cancer growth. Accordingly, tumor suppressors and oncoproteins are often analogized to the brakes, and accelerator, of a car (figure 19).
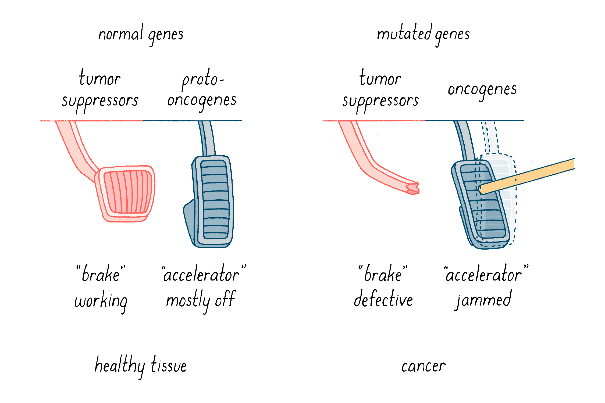
Figure 19: Tumor suppressors function as breaks on cell division, whereas, oncoproteins accelerate cell division. Mutations in tumor suppressor genes result in defective brakes, but mutations in oncogenes cause over acceleration (20).
In cancer, aberrant methylation of CpG islands can lead to reduced production of tumor suppressors and/or increased production of oncoproteins, resulting in abnormal cellular proliferation. Abnormal hypermethylation of tumor suppressor gene promoters manifests as reduced tumor suppressor protein production, and a loss of the brakes on cell proliferation (figure 20). Aberrant hypomethylation of oncogene promoters can cause increased oncoprotein production and acceleration of cell proliferation.
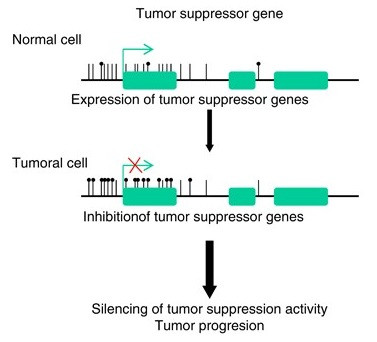
Figure 20: Tumor suppressors function as breaks on cell division in normal cells (top); the promoter is largely unmethylated (lack of black circles). In cancer cells, abnormal hypermethylation (black circles) of tumor suppressor gene promoters can result in reduced tumor suppressor gene expression (bottom-red X), manifesting as unchecked cell proliferation and tumor progression (21).
During oncogenesis hundreds of genes are silenced or activated via methylation. Presently, there are two hypomethylating agents that inhibit DNA methyltransferase methylation of cytosine residues in the clinic, azacytidine and decitabine (figure 21), used to treat MDS, AML, CMML, JMML, etc.
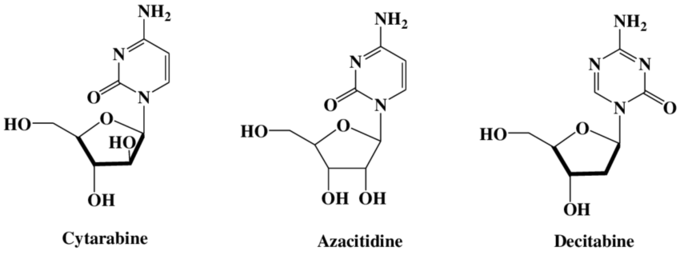
Figure 21: Azacitidine and decitabine are hypomethylating agents that play central roles in the treatment of several hematologic malignancies, including AML, MDS, JMML, CMML, etc. (22).
Differential Methylation
Regions of DNA, despite being identical in sequence, can be uniquely methylated in different tissues (figure 22).
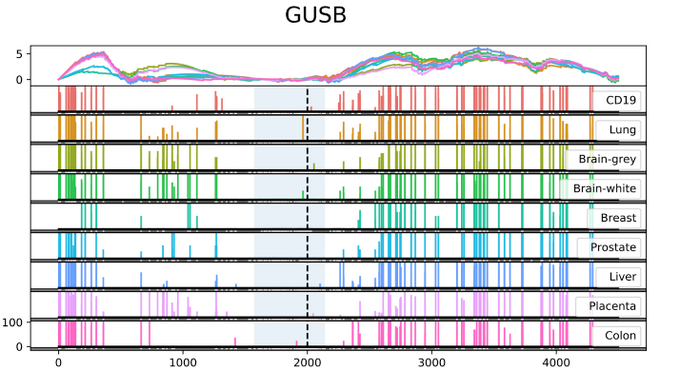
Figure 22: Differential methylation of the GUSB gene between tissues. Each vertical bar corresponds to the degree of methylation evident at the corresponding nucleotide (x-axis). The blue-shaded area corresponds to a CpG island; please note it is largely unmethylated (23).
Similarly, malignancies can have unique DNA methylation signatures depending on what tissue they originated from (figure 23).
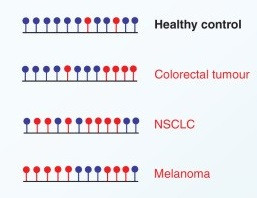
Figure 23: Differential methylation of the same region of DNA in normal tissue and various malignancies. Red circles correspond to unmethylated cytosines, whereas, blue circles are methylated cytosines (24).
Detecting DNA Methylation in Cancer Screening
DNA methylation can be detected in numerous ways, including mass spectrometry, methylation specific PCR (MSP), whole genome bisulfite sequencing, methylation specific bisulfite-Seq (MSBS), Illumina Methylation Assay, etc. The primary principle underlying these methods is the use of sodium bisulfite which converts unmethylated cytosines of CpG dinucleotides to uracil, but does not alter methylated cytosines (figure 24).
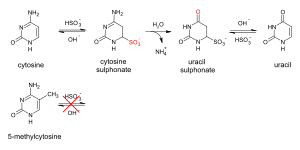
Figure 24: Sodium bisulfite converts unmethylated cytosines of CpG dinucleotides to uracil, but methylated cytosines are unaffected (25).
Polymerase chain reaction (PCR) or next generation sequencing (NGS) are often used after sodium bisulfite treatment to determine the methylation state of the tissue tested (figure 25). Many genomics companies use differential methylation techniques to determine if a patient has cancer and where it originated from. We will discuss this in more detail later.
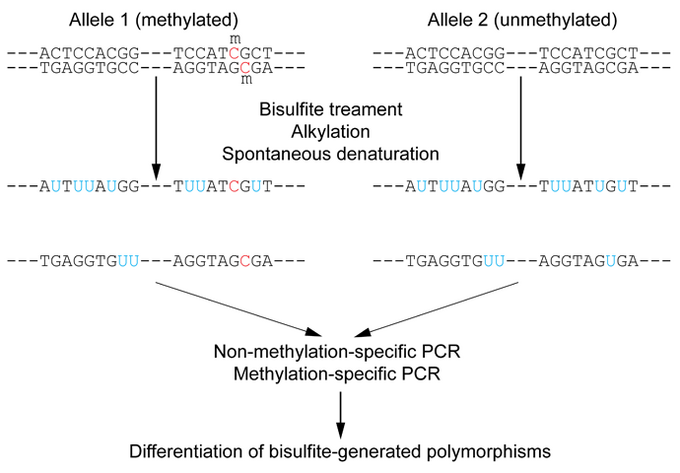
Figure 25: Sodium bisulfite converts unmethylated cytosines of CpG dinucleotides to uracil, but methylated cytosines are unaffected. PCR, NGS, etc., are then used to characterize the differential methylation of the sample tested (26)
The aforementioned discussion of bisulfite sequencing is intentionally over simplified. In reality, it is far more nuanced and is prone to errors due to DNA degradation, incomplete bisulfite processing, etc.
Liquid Biopsy
Liquid biopsy refers to the molecular analysis of cfDNA, RNA, subcellular structures, proteins, circulating tumor cells, etc., in biological fluids (figure 26). It is a catch all term referring to tests that enable the diagnosis, or analysis, of tumors using fluid samples, including blood, cerebrospinal fluid (CSF), saliva, etc. However, we generally use the term liquid biopsy to be synonymous with sampling of blood, as opposed to other sources. In this article we will focus on cfDNA, but in future articles we will explore many other facets of liquid biopsies through our exploration of multiomics.
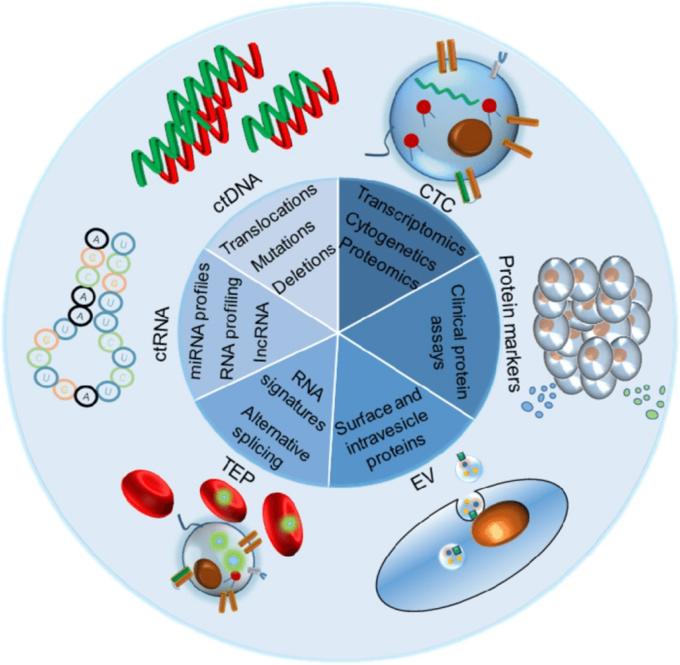
Figure 26: Liquid biopsy markers used in cancer testing. EV=extracellular vesicles, TEP=tumor-cultured platelets, ctRNA=circulating tumor RNA, ctDNA=circulating tumor DNA, CTC=circulating tumor cells (27).
cfDNA
cfDNA refers to DNA released by cells into biologic fluids. It was first identified in 1949 when cellular DNA derived from normal cells was isolated from the blood of patients. It is now established that many different types of cells release their DNA into biologic fluids, including cancer, fetal, and normal cells.
cfDNA is released from cells by three principal processes, including apoptosis, necrosis, and secretion of exosomes (figure 27).
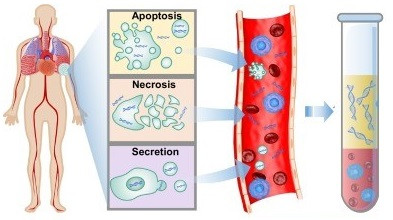
Figure 27: cfDNA is principally generated by apoptosis, necrosis, or exosome secretion (28).
cfDNA Derived from Apoptosis
Apoptosis, also termed “cellular suicide”, is the process of programmed cell death that occurs normally during development and aging to maintain cellular homeostasis. It is a highly regulated process that can be activated by various pathways beyond the scope of this article, and results in the death of 50 to 75 billion cells daily in average adults. Apoptosis is considered a vital facet of normal development, immune system function, etc., as it allows for the removal of senescent or obsolete cells without inducing inflammation. Apoptosis is comprised of a series of cellular morphological and biochemical changes, including pyknosis, chromatin condensation, DNA and nuclear fragmentation, cell membrane remodeling and blebbing, and apoptotic body formation (figure 28).
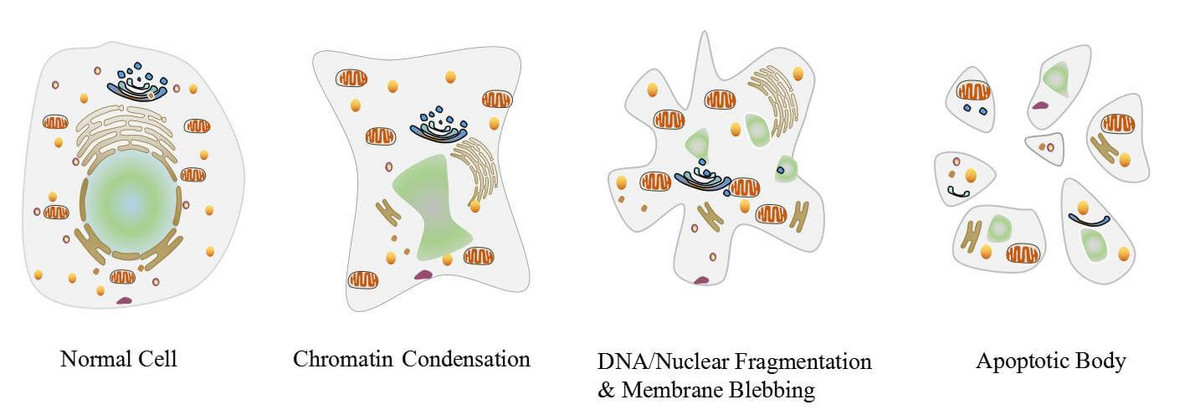
Figure 28: Apoptosis is a highly regulated process of programmed cell death resulting in chromatin condensation, DNA and nuclear fragmentation, membrane blebbing, and apoptotic body formation (29).
Usually, the remnants of apoptosis are phagocytosed by macrophages. However, some apoptotic debris is detectable in the blood, including cfDNA. The cfDNA generated by apoptosis, unlike necrosis (see below), is usually 160-180 base pairs with a predominant peak of 166 base pairs. This corresponds to the 147 DNA base pairs usually associated with histones in the nucleosome and the associated linker DNA that bridges nucleosomes. This important characteristic of apoptosis related cfDNA is capitalized on by current liquid biopsy methods, which we will discuss later.
cfDNA Derived from Necrosis
Whereas apoptosis is a highly regulated, programmed, physiologic process critical to cellular homeostasis, necrosis is characterized as pathologic premature cell death triggered by trauma, infection, cancer, ischemia, toxins, etc. Apoptosis, unlike necrosis, is a systematic process that predictably manifests as DNA and nuclear fragmentation, membrane blebbing, and apoptotic body formation, and doesn’t classically induce inflammation. In contrast, necrosis results from catastrophic disruption of cell membrane homeostasis and deregulation of sodium/potassium pumps. This culminates in membrane disruption and blebbing, mitochondrial and cytoplasmic swelling (oncosis), nuclear disruption (karyolysis), and associated inflammation (figure 29). DNA released via necrosis tends to be much longer than apoptosis, often larger than 10,000 base pairs.
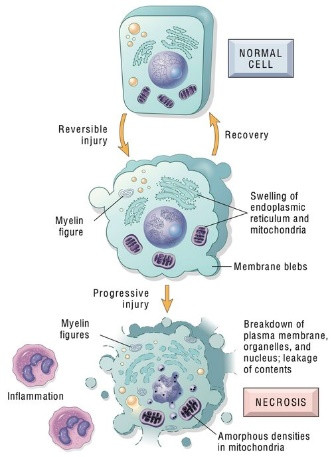
Figure 29: Necrosis is induced by numerous cellular insults and manifests as a breakdown of the cellular plasma membrane, nucleus, etc., and haphazard leakage of cellular contents into biologic fluids (30)
cfDNA Derived from Cell Exosome Secretion
Nearly all human cells release extracellular vesicles (EVs) that are lipid-bilayer-encapsulated parcels carrying a diversity of molecular cargo. Exosomes are small EVs of 30-100 nms (figure 30). They often contain carry cfDNA fragments ranging between 150 and 6000 base pairs. Accordingly, exosomes have been implicated in the transfer of genetic material from one cell to another. Regardless, although much remains unknown about exosomes, their presence in biologic fluids undoubtedly facilitates isolation of cfDNA for testing.
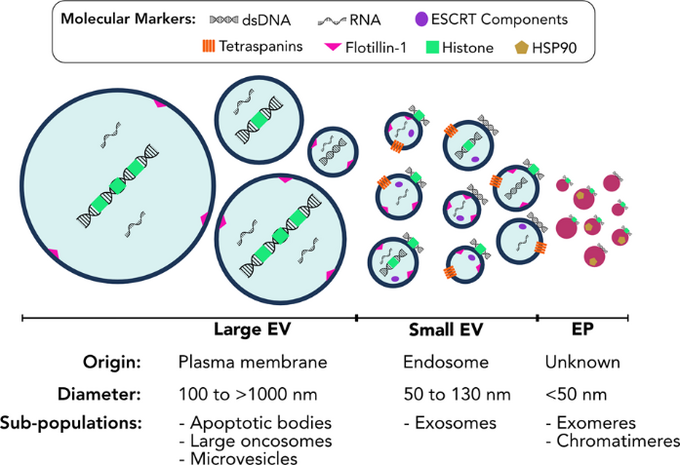
Figure 30: There are numerous types of extracellular vesicles (EVs). Exosomes, derived from endosomes, are small EVs usually 30-100 nms (31).
Other Sources of cfDNA
As aforementioned, the three predominant sources of cfDNA are apoptosis, necrosis, and cellular exosomes. However, cfDNA also includes mitochondrial DNA (40-300 bps), NETosis related DNA, protein complexed DNA, extrachromosomal circular DNA (30-20,000 bps), etc. (27). Indeed, pyropoptosis, mitochondrial catastrophe, autophagy, phagocytosis, and active cellular release of macromolecules and microvesicles (1000-3000 bps), can liberate cell DNA into biological fluids (figure 31). These topics are beautifully reviewed elsewhere (27), and beyond the scope of this discussion.
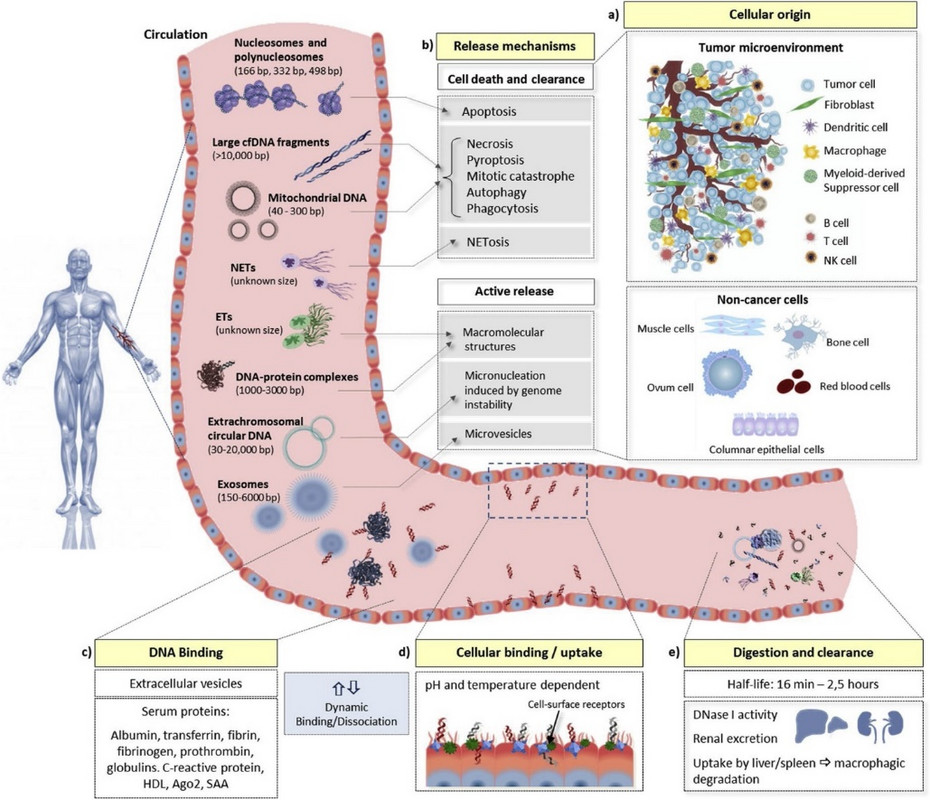
Fig. 31: Characteristics of cell-free DNA in the human body. (a) In cancer patients, cell-free DNA (cfDNA) originates from multiple sources, including cancer cells, cells from the tumor microenvironment, and non-cancer cells from other regions of the body (e.g., hematopoietic stem cells, muscle cells, and epithelial cells). (b) DNA can be liberated from these cells via different mechanisms, most prominently apoptosis, necrosis, and active secretion, although other forms of cell death and clearance may contribute. The physical characteristics of the cfDNA produced by these different mechanisms vary considerably. cfDNA levels are further influenced by its (c) dynamic association and disassociation with extracellular vesicles and several serum proteins, (d) rate of binding, dissociation and internalization by cells, which is dependent on pH, temperature, and can be inhibited by certain substances (such as heparin), and (e) the rate of digestion or clearance, including the activity of DNAse I, renal excretion into urine, and uptake by the liver and spleen (27).
The Clinical Utility of cfDNA
cfDNA is released by many types of cells, including healthy, fetal, and cancer cells (figure 32). Accordingly, cfDNA is used in obstetrics to identify genetic abnormalities in the fetus. It’s used in solid organ and bone marrow transplant to assess for rejection, and donor chimerism, respectively. And it’s used in cancer patients in multiple capacities we will discuss soon.
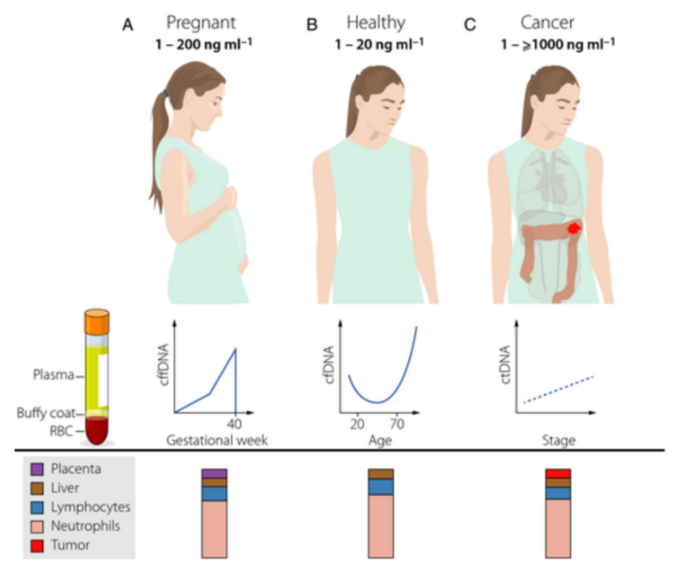
Figure 32: Circulating fetal (cffDNA), healthy (cfDNA), and tumor (ctDNA) can be found in cell-free DNA (32).
The concentration of cfDNA in the plasma of healthy individuals is usually 1-20 ng/ml, and fluctuates with age (32). The majority of healthy cfDNA emanates from hematopoietic cells, including white blood cells (55%), and erythrocyte (red blood cell) progenitors (30%). About 10% of cfDNA arises from endothelial cells and 1% from hepatocytes (liver cells). Other cells contribute small amounts of DNA to the overall cfDNA population (figure 32).
The amount of cell free fetal DNA (cffDNA) increases during pregnancy, and is presently employed to assess for fetal genetic aberrations (figure 32). However, the FDA recently encouraged caution when using tests as many of them are not FDA approved.
cfDNA is generally more abundant in cancer patients than healthy individuals, as cancer cells can release copious amounts of cfDNA, depending on the tumor type, stage, histology, etc. cfDNA originating from tumor cells is called circulating tumor DNA (ctDNA). In patients with late-stage tumors, ctDNA can be up to 1000 ng/ml, far in excess of the typical 1-20 ng/ml of normal cell. To this end, cfDNA originating from non-malignant cells coexists with tumor cell derived ctDNA in the total cfDNA of cancer patients. Thus, ctDNA is present in circulation as a fraction of total cfDNA in patients with cancer, and can encompass 0.01% to 90% of total cell-free DNA, depending on the patient’s cancer type, stage, etc.
The ratio of ctDNA to noncancerous DNA drastically impacts the sensitivity of liquid biopsy for malignancy. Small amounts of ctDNA, frequently seen in early-stage malignancies, can be difficult to accurately detect. In contrast, it’s generally much easier to identify late-stage cancer in patients due to the profound amount of ctDNA in their blood (figure 32).
Clearance of cfDNA
cfDNA and ctDNA is generally cleared from the circulation with an estimated half-life of 15 minutes to 2.5 hours (27). The mechanisms whereby cfDNA, including ctDNA, is cleared from the blood are poorly understood, but renal excretion, hepatic uptake, and the spleen have been implicated in this process. Regardless, the relatively rapid clearance of ctDNA from the circulation makes assessment of ctDNA extremely useful in identifying residual cancer post-treatment.
Liquid Biopsy Revisited
In 1989, tumor DNA circulating in the blood was first identified. Since then, many companies have strived to develop liquid biopsies; blood-based tests that allow for early cancer detection, identification of cancer cell gene mutations, prediction of treatment response, etc.
The ability to simply obtain a sample of blood from a patient to determine if they have cancer and how they will respond to various treatments is incredibly appealing. When compared to the traditional approach of biopsying a patient’s tumor, liquid biopsy is much less invasive, safer, cheaper, quicker to obtain, more reliable, without sampling error, often times more instructive of a tumor’s heterogeneity, better at rapidly delineating response to treatment, etc.
Numerous companies now specialize in liquid biopsy, and many others intend to. The field, although in its relatively infancy, is already saturated and will become much more so in the future. The reason for this is that liquid biopsy currently constitutes a roughly 9.2-billion-dollar industry, but is expected to grow to 26.2 billion by 2030 (figure 33). Accordingly, Exact Science purchased Thrive for 2.5 billion dollars. Illumina purchased GRAIL, itself an Illumina spinoff, for 8 billion dollars. Roche now has 360 million dollars invested in Freenome. To this end, my expectation is that liquid biopsy will become remarkably ubiquitous in the relative future, as patients will have annual cancer screenings via a simple blood test at their yearly medical check-ups.
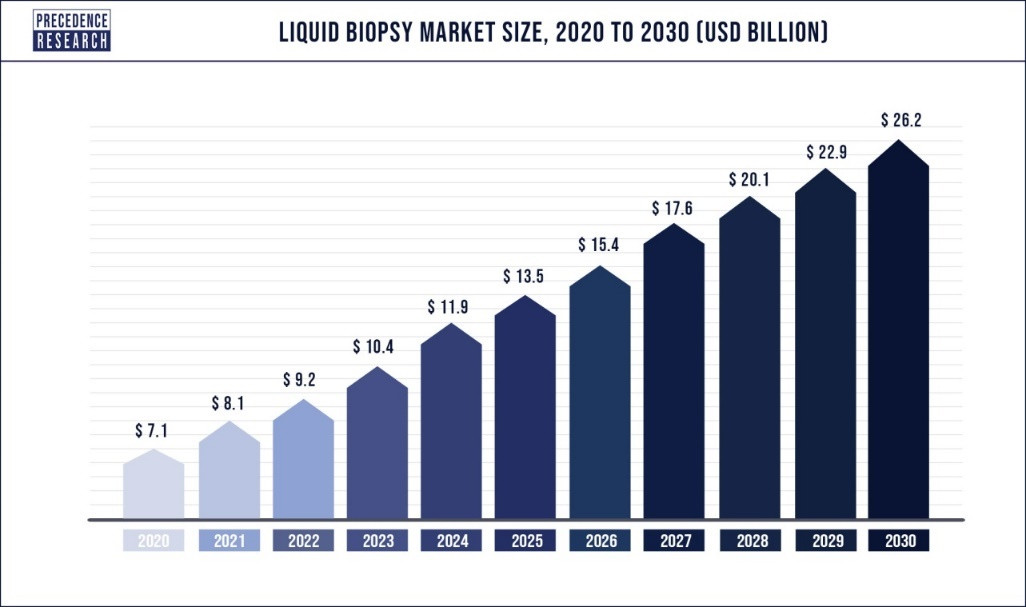
Figure 33: The liquid biopsy market is expected to grow significantly (33).
It's important to recognize that liquid biopsy is generally used to describe the analysis of cfDNA present in blood to delineate a patient’s cancer-specific information. However, in the strictest sense, liquid biopsy can involve the sampling of any biologic fluid (figure 34), including saliva, blood, CSF, urine, sputum, etc., for circulating protein, DNA, RNA, and circulating tumor cells (CTCs).
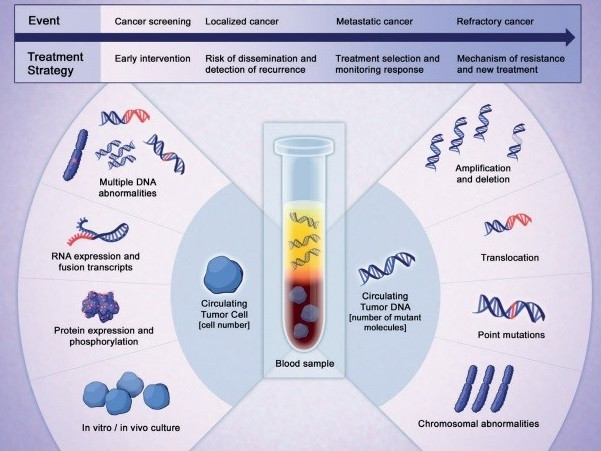
Figure 34: Liquid biopsy applies to the sampling of RNA, proteins, DNA, etc., in a patient’s biological fluid (34).
Multiomics in Liquid Biopsy
Multiomics is a relatively new approach where data sets obtained from different “omic” groups are combined to analyze samples of interest. This includes proteomics, RNAomics, genomics, transcriptomics, epigenomics, lipidomics, glyooimics, metabolomics, microbiomics, etc. (figure 35). Many of these, and multiomics itself, is the focus of future entries in this series.
Figure 35: Multiomics includes, metabolomics proteomics, genomics, epigenomics, etc. (35).
Nonetheless, it’s imperative to understand that liquid biopsy companies often employ different multiomic strategies predicated on identifying some variation of circulating protein, DNA, RNA, CTCs, etc. The specific strategy they use is a key differentiator, and determinant of the efficacy of their assays. We will spend considerable time exploring this in our comparative analysis of the respective companies in Chapter 2 of this mini-series on cfDNA.
From Blood Draw to Diagnosis
Liquid biopsy begins with phlebotomy at the bed side and ends with high-level characterization of a patient’s cancer or treatment response, presence of malignancy, etc. In identifying if a patient has a cancer or not, the ideal liquid biopsy test would be 100% sensitive and 100% specific, with an area under the curve (AUC) of 1. This represents a test that identifies every patient with cancer as having cancer, and every patient without cancer as not having cancer. Accordingly, the primary measure of a liquid biopsy is its sensitivity, specificity, and AUC. This is fundamental to the liquid biopsy company comparative analysis we will perform in Chapter 2.
The Process
To perform liquid biopsy a patient’s blood is drawn in a tube containing EDTA to prevent heparin related DNA degradation (figure 36). Plasma is separated from the remainder of the blood via centrifugation. DNA extraction to obtain plasma cfDNA in the blood and extracellular vesicles is usually performed via silica-based methods based on the reversible interaction between the carboxyl surface of silica and DNA. Negative charges on silica surfaces and DNA backbones are bridged by divalent cations, facilitating binding of DNA to silica. DNA is eluted from silica by water which precludes the interaction of silica with DNA. One can isolate specific lengths of cfDNA by choosing different silica-based extraction methods. For example, silica coated magnetic beads recover shorter cfDNA fragments (<600 bps), whereas silica column-based methods extract longer cfDNA fragments.
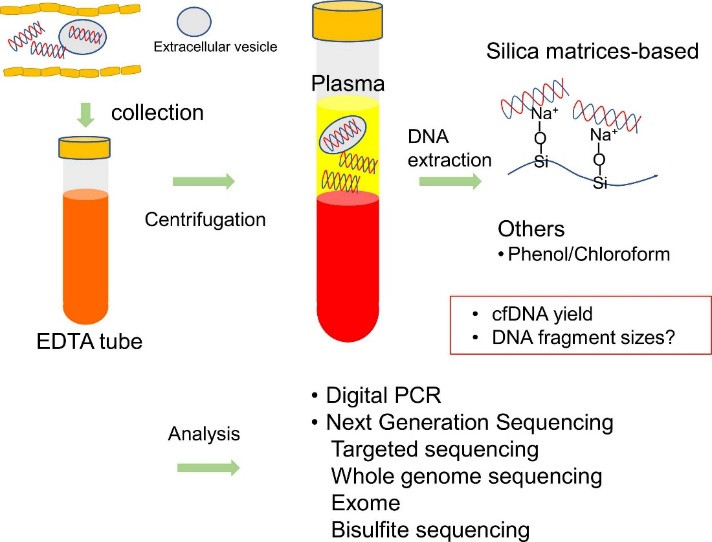
Figure 36: High-level overview of cfDNA testing (27).
Subsequent to cfDNA extraction via silica, proteinase K treatment and phenol/chloroform extraction removes proteins. DNA can be recovered by ethanol precipitation. Of course, there are many methods for isolating cfDNA from plasma, and each method can preferentially isolate DNA fragments of certain sizes with different yields. This is critical as single-nucleosome sized fragments, usually around 166 base pairs, have been shown to be enriched with tumor specific mutations compared to longer DNA. This suggests DNA extraction techniques that isolate shorter DNA fragments are more optimal than others for identifying cancer. Once cfDNA has been isolated, there are two principal approaches to evaluating it; single-gene and genomic profiling analysis.
One commonly used single-gene based approach to cfDNA is digital polymerase chain reaction, such as BEAMing and droplet digital PCR. It is the most current PCR technique with superior sensitivity and specificity used for cfDNA analysis. However, it mandates that one knows the cancer specific gene mutations of interest, and requires pre-designed TaqMan probes for these target genes. This technique offers excellent sensitivity as variant alleles of 0.1% frequency can be detected. Droplet digital PCR is a single-gene based approach presently used to frequently characterize EGFR and PIK3CA mutations.
Genomic profiling analysis often utilizes next generation sequencing (NGS) based assays that detect mutations in cancer related genes without prior knowledge of the mutations, as opposed to targeted sequencing of gene panels (figure 37). NGS approaches require enrichment of DNA fragments of interest from target gene loci before sequencing, either by PCR amplification of multiple primer sets or by hybridization-based capture. NGS has a random error rate of 0.1-1% in each platform, and can have difficulty confidently calling mutant alleles of frequencies of less than 1%. However, using unique molecular identifiers, random DNA sequences of 6-22 nucleotides, that tag DNA fragments before sequencing can facilitate detection of mutant alleles present at less than 1%. NGS-based FoundationOne liquid CDx test and Guardant 360 CDx for companion diagnosis are presently FDA approved.
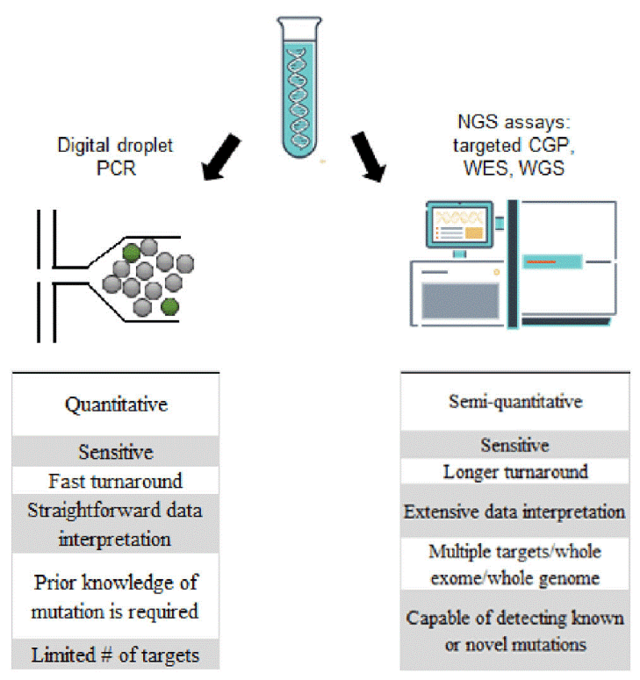
Figure 37: Comparison of digital droplet PCR and NGS (36).
NGS can detect copy number alterations. This is extremely useful as many cancers, such as MDS, AML, ALL, etc., can harbor alterations in chromosome copy numbers. However, alterations in copy number are difficult to delineate when the ctDNA tumor fraction is limited.
Differential DNA methylation is extremely useful in identifying if a patient has cancer, and if so, where it originated from. As we discussed earlier, cancer cells often have different methylation patterns than normal cells, and cells of different tissues have unique methylation signatures (figure 38). Accordingly, many liquid biopsy companies use bisulfite treatment of DNA, discussed in the section on DNA methylation testing, to discern the methylation pattern of extracted cfDNA. This is usually incorporated with aforementioned information about chromosomal copy number alterations, tumor specific mutations, other gene mutations, DNA fragment size, etc., in the full characterization of a patient’s cfDNA.
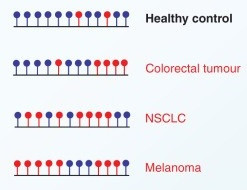
Figure 38: Differential methylation of the same region of DNA in normal tissue and various malignancies. Red circles correspond to unmethylated cytosines, whereas, blue circles are methylated cytosines (24).
Figure 39 illustrates the process whereby short DNA fragments are isolated and extracted from a patient’s biologic fluid. In this example, it’s then subjected to PCR based targeted sequencing to assess for tumor-specific gene mutations. This information is often interpreted in the context of parallel tests, such as DNA methylation identification, micro-RNA (miRNA) analysis, proteomics, etc., to yield a final assessment of a patient’s cancer status.
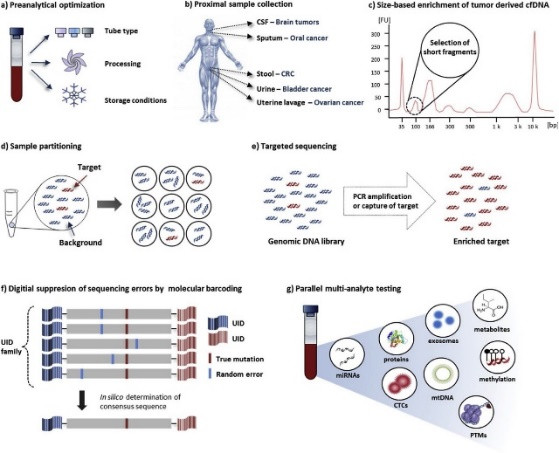
Figure 39: cfDNA from before drawing a patient’s blood to final analysis. Collection tubes (a) are prepared prior to a patient’s sample being drawn (b). cfDNA of desired length is identified (c) and isolated through sample partitioning (d). Thereafter, mutations of interest are detected by targeted PCR based sequencing (e), utilizing bar coding (f). Final analysis involves numerous tests done in parallel, which can include CTCs, methylation, miRNAs, etc. (24).
Although it may seem somewhat rudimentary, there is an art to cfDNA. The type of patient sample and how they are obtained, stored, transported, and processed has profound implications on the accuracy of cfDNA assays. Figure 40 illustrates how intricate this process can be.
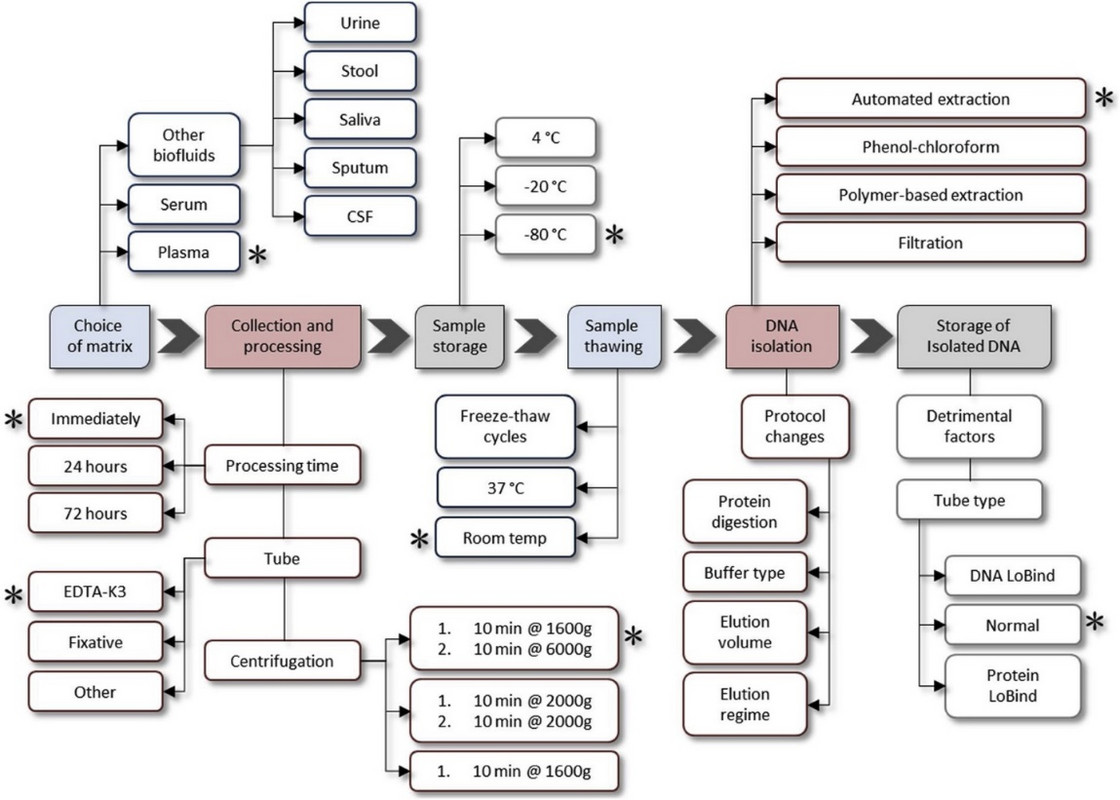
Figure 40: Overview of cfDNA testing revealing numerous intricacies (32).
Choose Your “Omics” Wisely
The choice of “omics” companies combine with cfDNA studies can provide a strategic advantage over their competitors if it leads to improved test sensitivity and specificity. Whether companies rely on isolating certain DNA fragment lengths, use differential methylation, perform whole exome sequencing via NGS or directed mutation mapping with PCR, etc., is a key differentiator between cfDNA tests themselves. In addition, which genes companies specifically assess in their cfDNA assays may give them an advantage over others. For example, if a company has identified a gene profile that better discriminates normal cells from cancer cells than other companies, this would be extremely advantageous.
Exact Science employs CancerSEEK in its population cancer screening. It was developed at Johns Hopkins University, and detects gene and protein biomarkers of cancer by probing 16 cancer genes, including PIK3CA, TP53, KRAS), and 9 clinically validated protein markers (CA125, CEA, etc.). Yet, the biggest player in the population cancer screening cfDNA industry now is arguably Illumina/GRAIL, although Guardant Health, Exact Sciences, Natera, Freenome, Singlear Genomics, etc., will have something to say about this, which we discuss in Chapter 2. Nonetheless, Illumina/GRAIL developed the Galleri test (figure 41) they contend can determine if a patient has one of 50 cancers from a single blood test (we will specifically review this in Chapter 2). Figure 41 shows the principles underlying GRAIL’s Galleri test.
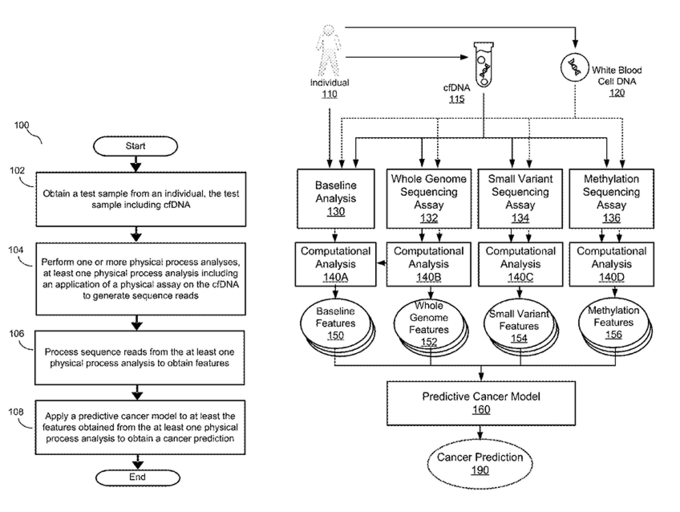
Figure 41: High-level overview of GRAIL’s Galleri test (37).
As you can see GRAIL’s test relies on whole genome sequencing, isolating and assessing for small genomic variants, and methylation sequencing.
For the individual that is unacquainted with genomics sequencing, AI/ML, cfDNA, etc., GRAIL’s technology appears daunting. However, for those familiar with these topics GRAIL’s methodology is actually fairly simplistic and appears to be easily imitated. Indeed, many companies employ similar techniques to assess cfDNA.
Consider the technology Freenome employs in figure 42. You can see significant similarities between what Freenome are GRAIL. However, Freenome specifically mentions RNA and protein analysis in their algorithm.
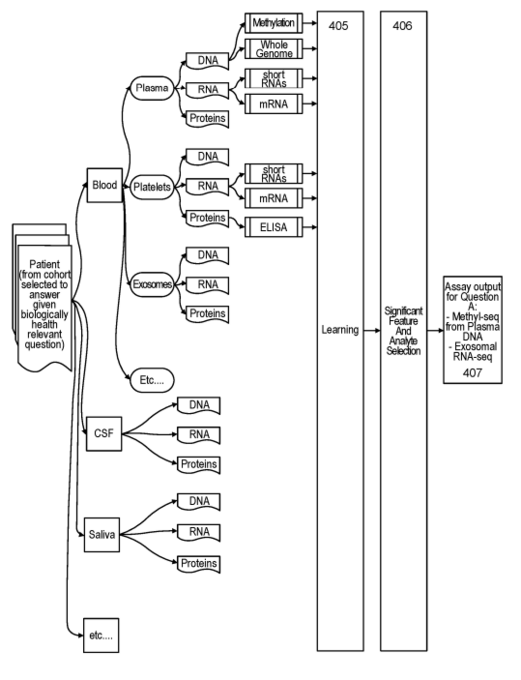
Figure 42: High-level overview of Freenome’s cancer screening paradigm (38).
Natera’s cfDNA algorithm is depicted below (figure 43). Importantly, as do numerous companies, Natera compares tumor DNA to germline DNA to delineate between cancer cell specific mutations and inherited ones.
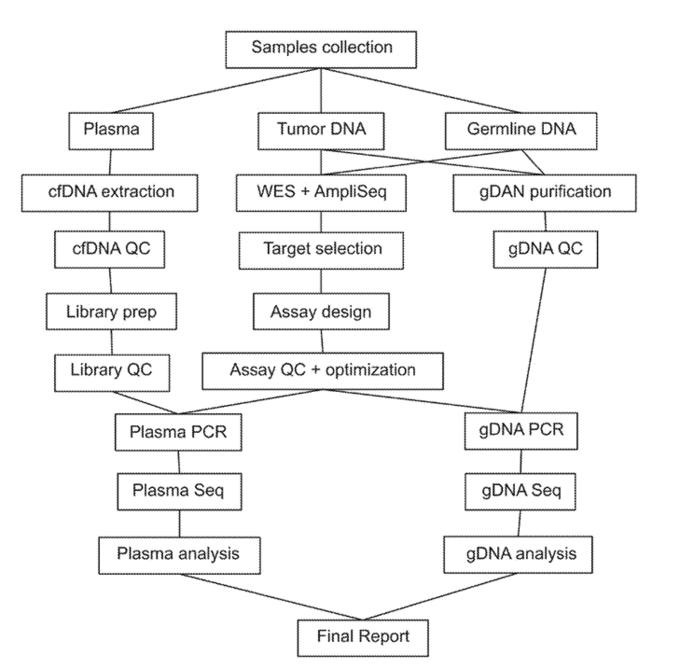
Figure 43: High-level overview of Natera’s cancer screening paradigm (39).
Ultimately, companies in the cfDNA space will distinguish themselves from one another based on the constitution of their assay, and their associated sensitivity, specificity, and AUC. We will do a deep dive on this in Chapter 2.
A Word on Artificial Intelligence/Machine Learning
There is no question the healthcare world will be dominated by AI/ML in the future (figure 44). Drug discovery, precision oncology, NGS, imaging, pathology, and patient data management have been inundated by AI/ML based healthtech companies. In fact, we analyzed several of these in the “Drowning in Data” article in this series. Path AI is FDA approved to assist pathologists with the diagnosis of prostate cancer. IDx-DR is an FDA approved retinal imaging device to detect diabetic retinopathy in diabetes. Skin Analytics just signed a contract with the UK National Health Service (NHS) to screen patients for melanoma using cell phones and their proprietary AI algorithm. Accordingly, I believe everyone should develop some familiarity with these disciplines, and I’ve provided a tremendous way to do so in the supplemental information 40).
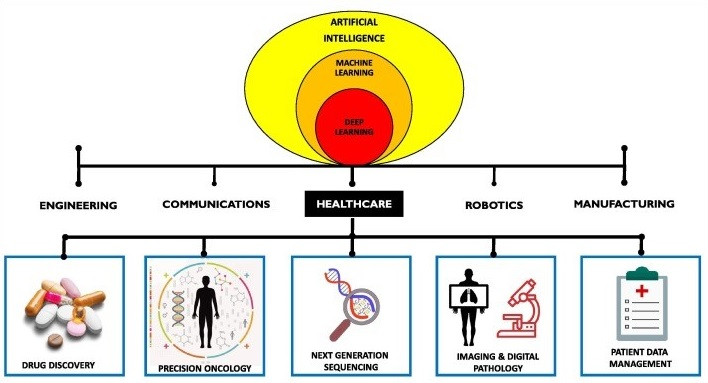
Figure 44: Applications of AI/ML in Healthcare (41).
Liquid biopsy companies in population cancer screening are tasked with identifying if a patient has cancer, generally with a single blood test. As such, their mission is to develop a model that can predict with high specificity and sensitivity if a patient has a malignancy based on the cell-free DNA and additional data they acquire from a patient’s sample. Accordingly, a cornerstone of cfDNA, and ultimately liquid biopsies, is the computational analysis performed to take the raw data obtained from a patient’s sample, including cfDNA assessment, to predict if they have cancer. This involves artificial intelligence (AI) and/or machine learning (ML) techniques. The fundamental premise of this is relatively simple, and involves supervised learning, as opposed to unsupervised learning.
Supervised learning trains a model using labeled data, whereas unsupervised learning uses unlabeled data (figure 45).

Figure 45: Supervised Learning trains a model using labeled data (left two panels), whereas unsupervised learning entails unlabeled data (right panel) (42).
For liquid biopsy models, supervised learning is required as a labeled data set corresponding to cfDNA tests matched with whether the patient had cancer or not is imperative (figure 46).

Figure 46: Supervised learning in cell-free DNA (cfDNA) studies requires a labeled data set including patients with cancer or not, and their corresponding cfDNA profile. Data sets are not usually as balanced as the one depicted here.
With this data set in hand liquid biopsy companies can begin developing their model for predicting if a patient has cancer or not based on their cfDNA. To do this, the data set is split into training, validation, and test sets. For simplicity we will include the test set in the validation set.
Often times, companies will use a 60:40, 70:30, 75:25, or 80:20 training:validation split of the data to train the model, and then validate it. Specifically, if one has 100 labeled cfDNA:cancer yes/no tests, often anywhere from 60-80 of those tests will be used to train the model, while the remainder will be used to validate it (figure 47).
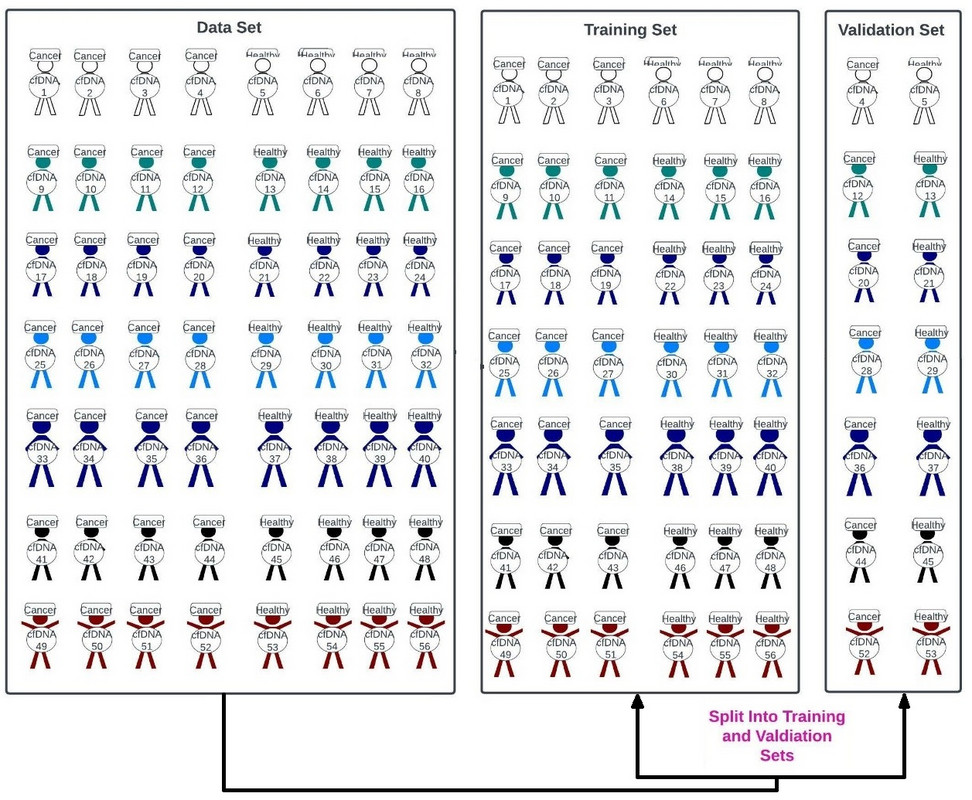
Figure 47: A 75:25 split of our data sets into training and validation sets.
The training set is used to train the model to identify a patient as having cancer or not based on their cfDNA profile (figure 48).
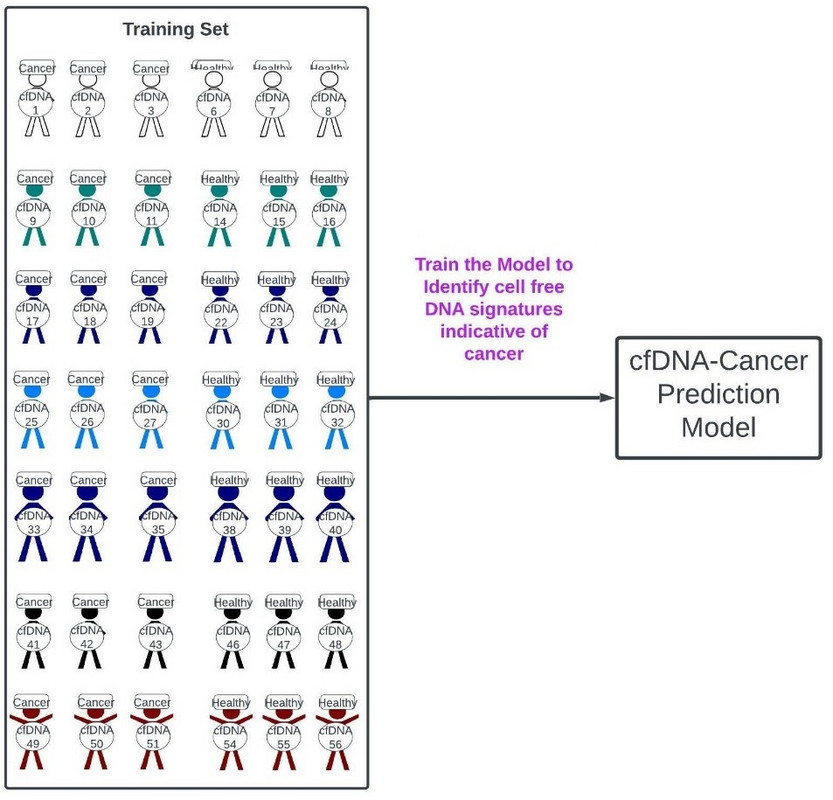
Figure 48: Training of our cfDNA-Cancer Prediction Model using the training data set.
The model is then used to predict whether or not patients in the validation set have cancer or not based on their cell-free DNA tests (figure 49). The model prediction is then compared to the known status of the cancer patient in the validation set to determine the accuracy, sensitivity (recall), precision, specificity, F1-score, AUC, etc., of the test (figure 49).
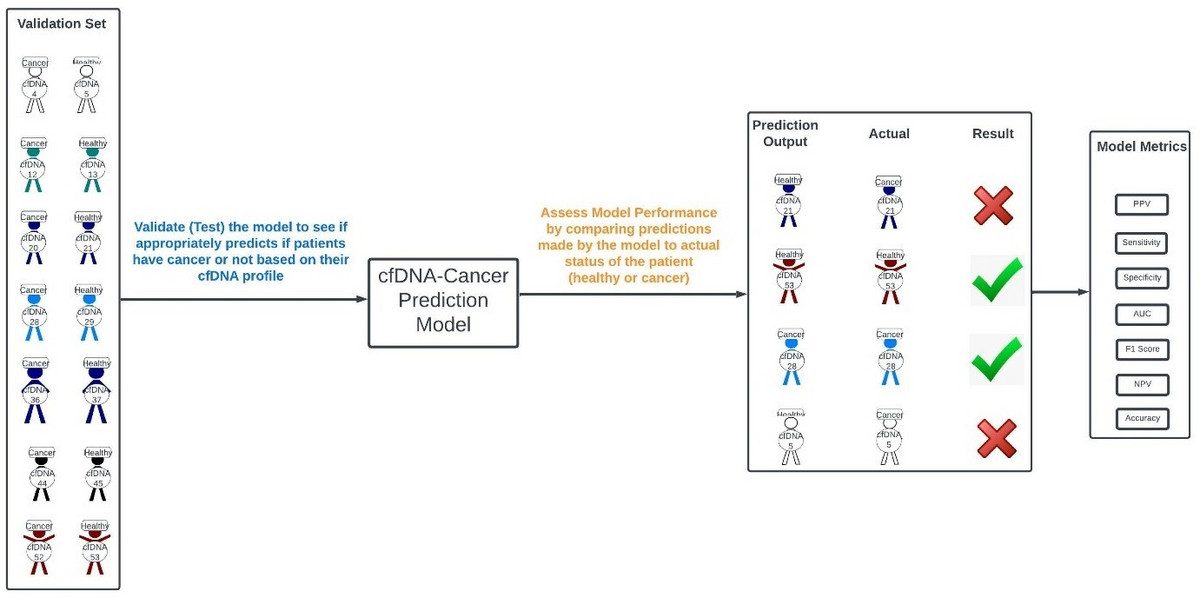
Figure 49: Testing of our cfDNA-Cancer Prediction Model using our validation set to determine if it appropriately identifies patients with cancer or not.
At this point we did a 75:25 training:validation split of our cfDNA:cancer yes/no data set. We trained our model by showing it the cfDNA results for our training set patients labeled as having cancer or not. Thereafter, we tested our model on the 25 remaining validation set patients to see if it appropriately identified them as having cancer or not.
The reality of cfDNA model development is far more complex than what I outlined. Nonetheless, the use of a training set and validation set pervades all of AI and machine learning, and the precision medicine forum.
A key concept of AI/ML is the optimization of the prediction model through reinforcement learning. Again, the underlying concept is simple but the real-world application is far more complex. Regardless, reinforcement learning, as it implies, involves refining a model through rewards and penalties. Specifically, in our case, if our model properly predicted a patient has cancer or not based on their cfDNA testing it’s rewarded. If it was wrong it’s penalized (figure 50), and the model is adjusted accordingly. The manner in which models are penalized or rewarded is beyond the scope of this paper.
Ultimately, after numerous model iterations predicated on penalties and rewards, the model that best predicts if a patient has cancer based on their cfDNA profile is identified (figure 50).
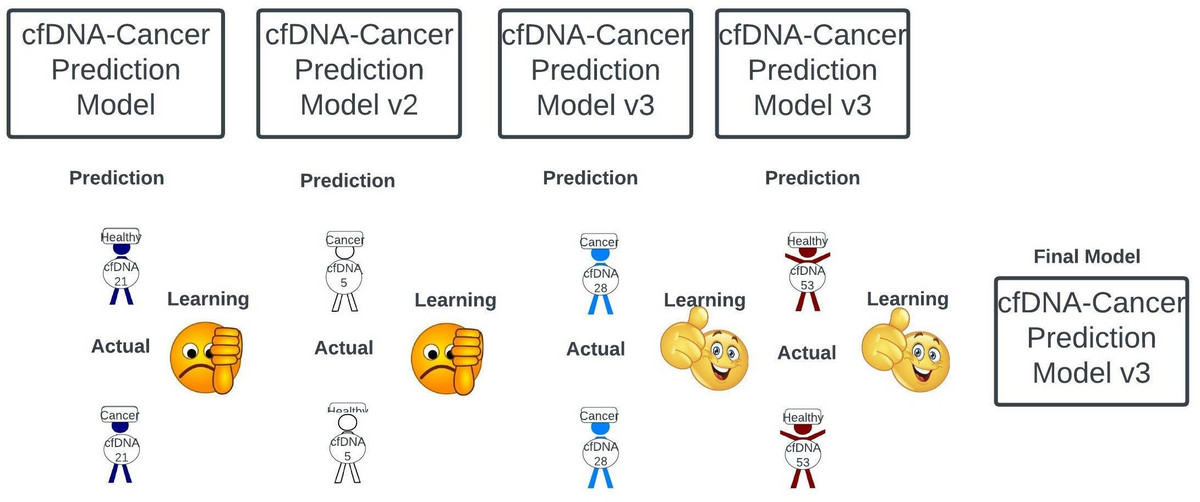
Figure 50: Reinforcement learning entails real-time optimization of our prediction model through. The model is penalized for making incorrect predictions (thumbs down), but rewarded when correct (thumbs up). Ultimately, after numerous model iterations, a final model is identified.
The finalized model is used to predict whether patients of unknown status, but known cfDNA profiles through liquid biopsy, have cancer or not (Figure 51). Patients identified as potentially having cancer are flagged, formally tested for malignancy, and treated if cancer is confirmed (figure 51).
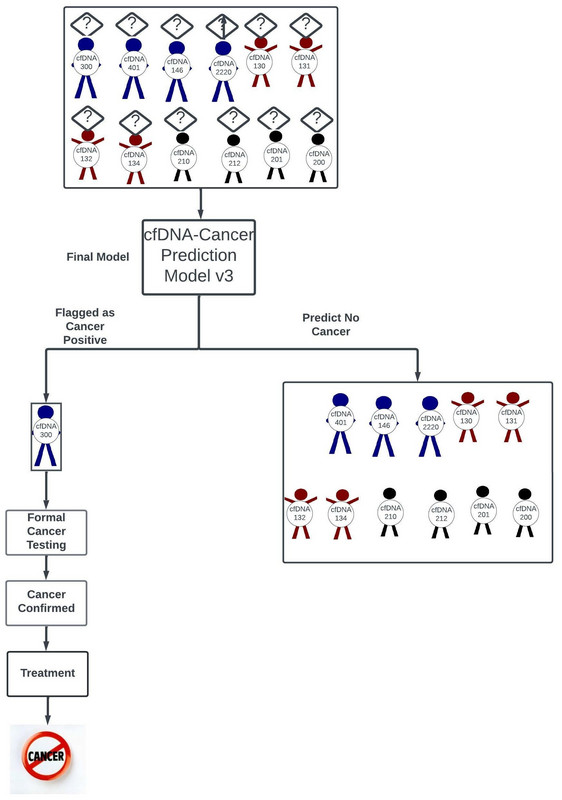
Figure 51: The finalized model is used to predict whether patients of unknown status, but known cfDNA profiles through liquid biopsy, have cancer or not. Patients identified as potentially having cancer are flagged, formally tested for malignancy, and treated if cancer is confirmed.
At the heart of every cfDNA population cancer screening company is their AI/ML algorithm for predicting if a patient has cancer based on their cfDNA profile. GRAIL lists this as “computational analysis” in their patent application (figure 52).
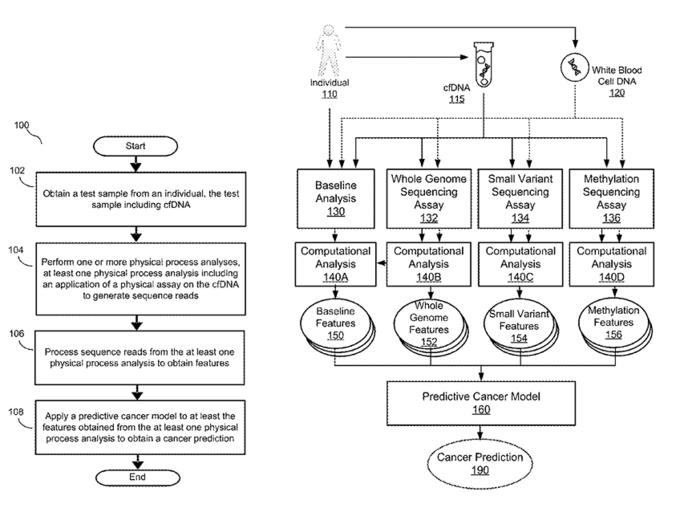
Figure 52: GRAIL’s depiction of computation analysis in their Galleri test paradigm (37).
Figure 53 illustrates Freenome’s patent application for their AI/ML liquid biopsy algorithm.
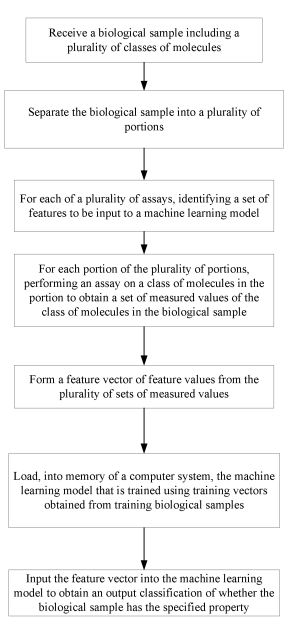
Figure 53: Freenome’s description of the way they generated their underlying ML prediction model (38).
There are numerous ways companies can build their AI/ML liquid biopsy algorithms, and this is another area where they can differentiate themselves from their competition. If their specific computational algorithm better predicts if a patient has cancer better than any of their competitors, it could be a huge financial boon for them. Accordingly, cfDNA companies invest tremendous resources in data science.
Data as the New Strategic Control Point
When one really looks under the hood of the use of cfDNA in medicine, it’s hard not to feel that it’s methodologically fairly simple. Performing the aforementioned DNA extraction, PCR or NGS, DNA methylation screens, etc., isn’t particularly complicated. Building the AI/ML algorithms theoretically isn’t that challenging. However, theory is very different from practice. Regardless, due to the overall theoretical simplicity of liquid biopsy, there has been an explosion of cfDNA companies over the last several years. I continue to read about new entrants in this space almost daily, despite Porter’s five forces for the industry becoming more daunting.
Ultimately, who wins in the liquid biopsy population cancer screening market should come down to two primary issues.
- How well do you identify patients with cancer as having cancer (sensitivity), and how well do you identify them as not having cancer when they don’t have cancer (specificity)?
- What is the cost-benefit ratio of your test?
Of course, there is far more to winning the liquid biopsy race, and we will review numerous strategic control points in Chapter 2, including the concept of customer service blueprinting.
Sadly, despite the aforementioned, the best cfDNA tests may not win the cfDNA population cancer screening race. Indeed, like everything else in life, marketing, politics, etc., will play a marked role here.
Illumina/GRAIL has brilliantly positioned itself for the future in the cfDNA population cancer screening space. They aggressively developed partnerships with numerous institutions, including the Cleveland Clinic, Dara-Farber Cancer Institute, Intermountain Healthcare, Mayo Clinic, Sarah Cannon, Sutter Health, US Oncology Network, Allegany Health System, Ohio State, Vanderbilt, Munich Re Life US, etc. If that wasn’t enough, GRAIL has a contract with the UK NHS to perform 140,000 of its Galleri cancer screening test, and an agreement with the US Department of Veterans Affairs for 10,000. These partnerships allow Illumina/GRAIL to tap into an insurmountable number of patients to not only test their Galleri assay and associated computational model, but to improve it. In fact, this might be the only example of a situation where a product can be optimized while it’s in clinical trials. Even if the test doesn’t work optimally the data Illumina/GRAIL will accumulate during testing will enable the company to make it better. In a world where data is king (figure 54), Illumina/GRAIL will be tough to conquer.

Figure 54: In the cfDNA race whoever has the data will likely prevail (43).
We will discuss the utility of GRAIL’s test across numerous cancers during Chapter 2. However, GRAIL is quickly becoming “too big to fail” despite not actually being FDA approved for population cancer screening. My hat is off to their marketing team as the world awaits the results of their studies.
Not to be out done Freenome has partnered with Biognosys, Brigham and Women’s Hospital, institutCurie, Qiagen, UC SanDiego Health System, UCSF, and the Parker Institute for Cancer Immunotherapy. Roche has 360 million dollars invested in Freenome.
Thrive, who Exact Science purchased for 2.15 billion dollars, has their CancerSEEK test that was developed at Johns Hopkins University, which we will dissect in Chapter 2. They have partnerships with Pfizer, Mission Wisconsin, etc., but seem to be lagging behind Freenome and GRAIL in this regard.
Ultimately, the more partners a cfDNA company has the more access they have to patients to refine their tests and algorithms. Moreover, just like anything in business, it’s much easier to capture new business than to steal it from a competitor. We will explore this in Chapter 2, particularly as we conceive of a way for our hypothetical company, Comprehensive Precision, to infiltrate the market even though there may be seemingly no more worlds left to conquer.
CfDNA in Cancer
We conclude Chapter 1 of our cfDNA discussion by discussing numerous potential applications of cfDNA in cancer (figure 55), including:
- Population Cancer Screening and Early Intervention
- Residual Disease Measurement
- Cancer Surveillance
- Predict Treatment Response
- Prognostication
- Account for Tumor Heterogeneity
- Dictate Treatment
- Identify New Drug Targets
- Drug Development
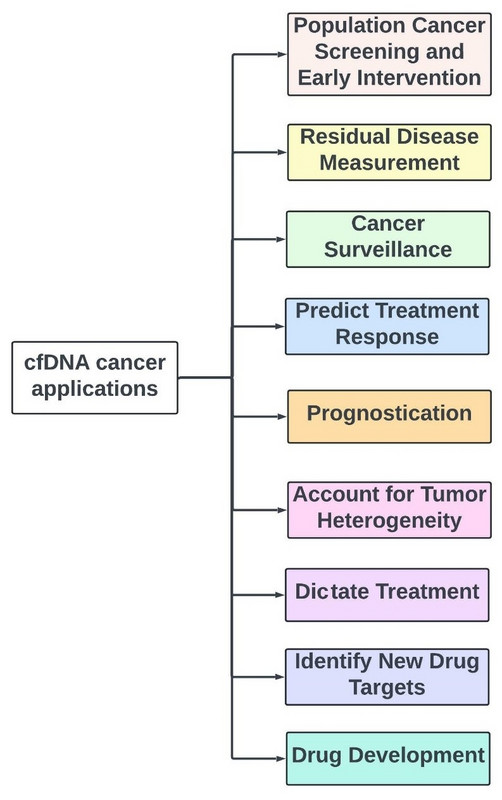
Figure 55: Utility of cfDNA in cancer care.
CfDNA in Population Health Screening and Early Intervention
The crux of the aforementioned discussion pertained to cfDNA in population cancer screening. There is little doubt in my mind there will be a day when cfDNA tests are performed annually in the clinic to determine if a patient has clinically and radiographically occult malignancy. This will facilitate early cancer interventions that could be life-saving. We will focus on this in Chapter 2.
Measuring Minimal Residual Disease
cfDNA is already being used to determine if patients have residual malignancy post treatment that is not radiographically or clinically apparent, referred to as minimal residual disease (MRD). To this end, it’s important to understand that a 1 cm cancer lesion on a CT scan constitutes 100 million cancer cells. Accordingly, a 1 million cancer cell lesion, amounting to 0.1 mms, would not be visualized on standard radiography. cfDNA will enable the clinician to determine if the patient has MRD, which may guide their treatment. Indeed, in ALL MRD evidence of persistent cancer prompts the oncologist to treat the patient with blinatumomab. In early June 2022, a study of cfDNA to guide the use of adjuvant chemotherapy in stage 2 colon cancer patients was conducted, the results of which were published in the NEJM. We will discuss this much more in Chapter 3.
Cancer Surveillance
There are numerous cancers where we don’t adequately assess for recurrent disease. For example, standard recommendations for non-metastatic breast cancer patients are to have physical examinations semi-annually, and breast imaging yearly even if they had advanced disease (e.g., stage 3). These patients would likely benefit from semi-annual cfDNA tests to identify early recurrence of their disease, assuming the tests meets the required sensitivity and specificity metrics. Indeed, this logic could be applied to almost every malignancy.
Predicting Treatment Response
It’s certainly conceivable that certain cfDNA signatures may correspond to patients responding to treatment, whereas others may confer resistance. Identification of such signatures may spare patients from receiving treatments that will not work, and ensure they receive those that will.
Our current immunotherapy prognostic measures are relatively crude, and involve testing microsatellite instability (MSI), tumor mutation burden (TMB), and PD-L1. If patients have high levels of any of these, they are more likely to respond to immunotherapy. However, many such patients don’t respond to immunotherapy. Moreover, patients that don’t have any of these characteristics sometimes respond to immunotherapy. One in 6 metastatic non-small lung cancer patients who were PD-L1 negative, MSI-low, and TMB-low respond positively to immunotherapy. cfDNA may yield an immunotherapy response signature one can use to better predict if patients will respond to immunotherapy, or any other treatment. Accordingly, numerous companies are in heated competition to develop clinically relevant immunotherapy prognostic assays, including Boston Gene, Oncohost, Oncocyte, etc.
Methodologically, using cfDNA to predict treatment response is easy to do, so long as one has a labeled data set of treatment responders and non-treatment responders. Accordingly, I contend that EVERY therapeutic clinical trial must incorporate cfDNA and/or other “omics” paradigms for optimal interpretation.
Prognostication
It's imperative to recognize no two tumors are created equal, even if they have the same stage and histology. Cancer is not binary and always resides on a continuum from indolent to aggressive. Even among generally aggressive metastatic melanomas there are some that grow slowly and are more amenable to treatment than others.
As per the “Quarterbacking a Patient’s Cancer Care” article (2), the best hematologists/oncologists are adept at delineating the underlying tumor biology. This is derived from observing the tumor's initial tumor burden, growth rate, response to treatment, molecular profile, histology, sites of metastases, etc. With every restaging scan performed after a designated treatment the oncologist learns more about the cancer and adjusts accordingly.
Imagine now if cfDNA was predictive of a tumor’s biology, and how it will behave and react to treatment. This would enable a physician to determine if they could “let up on the gas” or not, by holding treatment in a patient with stage 4 disease that is incurable. In fact, there are examples of stage 4 patients who were resistant to a given treatment that had their cfDNA go DOWN off treatment, and then respond favorably to the treatment when rechallenged (32).
Account for tumor heterogeneity
There is little we comprehend about the heterogeneity of tumor biology from patient to patient, and even in the same patient who has multiple metastatic sites of disease that respond differently to treatment. The 80-year-old female, depicted in figure 56, with metastatic triple-negative breast cancer (56a) was treated with three cycles of carboplatin/gemcitabine (56b). The right axillary lymph nodes and right pelvic lesion responded beautifully to treatment, but two new liver lesions developed. This mixed response to treatment, representing underlying tumor heterogeneity, prompted a change in treatment to sacituzumab, and is the bane of a oncologist's existence.
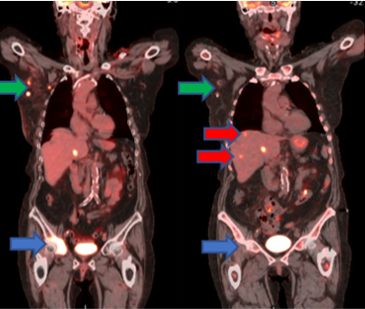
Figure 56: A patient with triple-negative breast cancer before (a) and after (b) three cycles of carboplatin/gemcitabine. The right axillary lymph nodes (green) and right pelvic lesion (blue) improved, but two new liver lesions (red) developed.
Historically, when we assessed the molecular composition of a tumor we obtained a tissue sample. We still do this, but liquid biopsies are frequently used now. One primary benefit of this is that liquid biopsies better account for tumor heterogeneity as tissue sampling only yields information about a solitary metastatic site, whereas liquid biopsy constitutes a simultaneous sampling of all disease sites.
Dictating Treatment
In my opinion there are two holy grails, no pun intended, for cfDNA:
- Population cancer screening
- Guiding treatment
At this time one of my biggest qualms with cell-free DNA, if I have any, is that I don’t know what to do with a patient who has no clinical or radiographic evidence of malignancy, but is cfDNA positive. Do I start treatment on that patient or wait? Does it impact overall survival? I suspect there will be a day in the not-too-distant future where cfDNA determines how we treat patients. In fact, the recent NEJM paper I alluded to earlier suggests we are nearly there.
Identifying New Drug Targets and facilitating drug development
cfDNA is in its relatively infancy right now. In the future these tests will expand to involve more of the genome, and be used in conjunction with multiomics. This will undoubtedly implicate numerous gene mutations we didn’t previously recognize as being oncogenic. This will facilitate the identification of new drug targets and drug development. This is exactly why Illumina is now moving into drug development, as I think every genomics company should be; Illumina recently announced an agreement with Vanderbilt to access their biobank for drug development purposes.
Tissue of Origin
As aforementioned, cfDNA will enable us to determine where tumors of unknown primary originate from, thereby enabling us to more effectively treat them.
Subclassification of Cancers
In the future it’s conceivable we will be subclassifying cancers based on findings from cfDNA patient testing. Entire cancer risk, staging, and treatment paradigms may be predicated on such discoveries.
Identification of Novel Inherited Mutations
There is little doubt that ubiquitous cfDNA testing of patients will precipitate the identification of novel hereditary mutations implicated in cancer formation. In addition, I believe cfDNA will unravel mutations that predispose patients to thrombosis. Indeed, I have numerous patients who develop blood clots for reasons we don’t understand. I suspect cfDNA will have a role to play here.
Chapter 1 comes to an end
After giving the aforementioned presentation to your board members as CEO of Comprehensive Precision, those that weren’t asleep give you a standing ovation. However, you quickly tell them to hold their applause, particularly as much of what you told them was predicated on incredible reviews written by individuals far better than you. Nonetheles, you’re well aware Chapter 1 was just the appetizer, and that the cfDNA main course and desert will be offered with Chapter 2 and Chapter 3, respectively, of the cell-free DNA series.
Conclusion
The “Insider’s Guide to Translational Medicine” editorial series has been tough to write. The subject matter we’ve attempted to tackle is vast and sophisticated. Moreover, the pace we’re going from one field to another has been breathtaking, even for me as the writer. To date, you bore witness to the FIRST EVER PRESENTATION of THE COMET ALGORITHM and CANCER PATIENT TREATMENT CARTOGRAPHY in the context of playing chess against cancer (1). We described the short-term approach to cancer care by being the FIRST PERSON EVER to describe how being a cancer doctor is similar to being an NFL quarterback (2). Moreover, we took an unprecedented dive into healthtech and how to interpret their contentions. Again, we introduced the world to A NEVER-BEFORE-SEEN BASIS method for assessing data and Harm/Inherency/Plan/Solvency/Disadvantage business rubric (3). Finally, we introduced cfDNA by catering to a remarkably heterogeneous audience, ranging from individuals who know nothing about DNA to those who do cfDNA for a living.
I hope the translational medicine tour we’ve taken under the umbrella of hematology/medical oncology has been eye opening for you. In the next entry in this series, Chapter 2 of our cfDNA expose, we will target population cancer screening. For what it’s worth, I will give my direct opinions on GRAIL, Freenome, Exact Sciences, Natera, GuardantHealth, etc. In addition, I will discuss the approach I would take as a new entrant to the space to try and garner market share in the presence of these talented Goliaths. I will introduce “customer service blueprinting”, and marketing principles such as segmentation, conjoint analysis, value chain, etc., in the context of cfDNA
I would love for you to join me. After all, you are the CEO of Comprehensive Precision.
References
- https://www.biopharmatrend.com/post/505-playing-chess-against-cancer-a-pharmaceutical-biotechnological-and-clinical-guide-to-modern-day-oncologic-treatment-cartography/
- https://www.biopharmatrend.com/post/510-quarterbacking-a-patients-cancer-care-for-submission/
- https://www.biopharmatrend.com/post/523-remember-to-be-precise/
- https://en.wikipedia.org/wiki/DNA
- https://en.wikipedia.org/wiki/Nucleobase
- https://nigms.nih.gov/education/fact-sheets/Pages/genetics.aspx
- https://www.nature.com/scitable/topicpage/translation-dna-to-mrna-to-protein-393/
- https://en.wikipedia.org/wiki/Chromatin
- https://www.genengnews.com/magazine/159/pcr-assay-for-chromatin-accessibility/
- https://www.genome.gov/genetics-glossary/Epigenetics
- https://www.internationaljournalofcardiology.com/article/S0167-5273%2814%2900361-1/fulltext
- https://link.springer.com/article/10.1007/s00018-021-03794-x
- https://en.wikipedia.org/wiki/Histone_acetylation_and_deacetylation
- https://en.wikipedia.org/wiki/DNA_methylation
- https://en.wikipedia.org/wiki/CpG_site
- https://upload.wikimedia.org/wikipedia/commons/0/0f/Cpg_islands.svg
- https://www.addgene.org/mol-bio-reference/promoters/
- https://en.wikipedia.org/wiki/DNA_methylation
- https://www.nature.com/articles/nrg.2016.83
- https://explorebiology.org/summary/genetics/the-genetic-basis-of-cancer
- https://www.researchgate.net/figure/Aberrant-DNA-methylation-patterns-in-human-cancer-Heavily-methylated-repetitive_fig3_51589121
- https://www.researchgate.net/figure/Chemical-structures-of-cytarabine-azacitidine-and-decitabine_fig16_272565621
- The Human Epigenomic Project
- https://www.researchgate.net/figure/Aberrant-DNA-methylation-patterns-in-human-cancer-Heavily-methylated-repetitive_fig3_51589121
- https://en.wikipedia.org/wiki/Bisulfite_sequencing
- https://en.wikipedia.org/wiki/Bisulfite_sequencing
- Abel Jacobus Bronkhorst, Vida Ungerer, Stefan Holdenrieder. “The emerging role of cell-free DNA as a molecular marker for cancer management”. Biomolecular Detection and Quantification, Volume 17, 2019.
- https://www.cell.com/trends/molecular-medicine/fulltext/S1471-4914%2821%2900002-2
- https://microbenotes.com/apoptosis/
- https://theartofmed.wordpress.com/2015/05/29/pathologic-cell-injury-and-cell-death-ii-necrosis/
- https://exosome-rna.com/bioactive-dna-from-extracellular-vesicles-and-particles/
- M. Cisneros-Villanueva, L. Hidalgo-Pérez, M. Rios-Romero, A. Cedro-Tanda, C. A. Ruiz-Villavicencio, K. Page, R. Hastings, D. Fernandez-Garcia, R. Allsopp, M. A. Fonseca-Montaño, S. Jimenez-Morales, V. Padilla-Palma, J. A. Shaw, A. Hidalgo-Miranda, Cell-free DNA analysis in current cancer clinical trials: a review, British Journal of Cancer, 10.1038/s41416-021-01696-0, 126, 3, (391-400), (2022).
- https://www.precedenceresearch.com/liquid-biopsy-market
- https://www.gene-quantification.de/liquid-biopsy-index.html
- http://www.biomech.ulg.ac.be/project/multi-omics/
- Ramkissoon, Lori & Pegram, Worthy & Haberberger, James & Danziger, Natalie & Lesser, Glenn & Strowd, Roy & Dahiya, Sonika & Cummings, Thomas & Bi, Wenya Linda & Abedalthagafi, Malak & Sathyan, Pratheesh & McGregor, Kimberly & Reddy, Prasanth & Severson, Eric & Williams, Erik & Lin, Douglas & Edgerly, Claire & Huang, Richard & Hemmerich, Amanda & Ramkissoon, Shakti. (2020). Genomic Profiling of Circulating Tumor DNA From Cerebrospinal Fluid to Guide Clinical Decision Making for Patients With Primary and Metastatic Brain Tumors. Frontiers in Neurology. 11. T1 - Cell-free circulating tumor DNA (ctDNA) in metastatic Renal Cell Carcinoma (mRCC): Current Knowledge and Potential Uses
- (private file reference)
- US Patent application PCT No: PCT/US2019/027565
- Natera US Patent Application
- https://www.mygreatlearning.com/mit-data-science-program
- Zodwa Dlamini, Flavia Zita Francies, Rodney Hull, Rahaba Marima. Artificial intelligence (AI) and big data in cancer and precision oncology. Computational and Structural Biotechnology Journal, Volume 18, 2020, Pages 2300-2311
- https://livebook.manning.com/book/grokking-machine-learning/2-1-what-is-the-difference-between-labelled-and-unlabelled-data-/v-4/
- https://www.linkedin.com/pulse/data-king-one-who-masters-rule-world-so-you-ready-vinay-rao-mle-/



{kind=link}