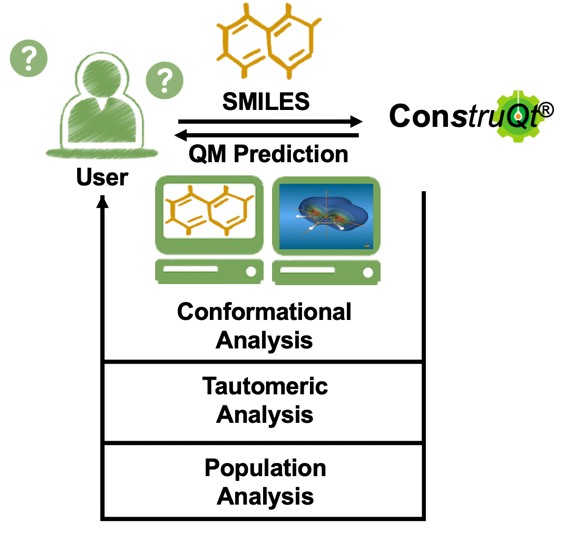
A Reliable Molecular Structure Predictor in the Cloud
Since August Kekulé’s proposal for the tetrahedral configuration of carbon or his more famous realization that benzene was a cyclic molecule, a snake biting its tale, molecular structure has been the leading consideration for the design of new molecules as drugs or performance materials. For the former, it is said that 70% of drug design is based on molecular shape with the remainder attributed to electrostatic or non-bonded interactions.
Structural chemistry began around the 1860 with these dual assignments by Kekulé but it wasn’t until one hundred years later with Allinger’s initial force field approaches that the first classical molecular mechanics (MM) models became available to make computer-assisted prediction of molecular structure. These models themselves are based on principles derived by Robert Hooke, a contemporary of Isaac Newton, in the mid 17th century with additional layers from van der Waals (19th century) etc.
The application of modern physics in the form of the quantum theory / mechanics (QM) developed in the first half of the 20th century by Schroedinger, Dirac and others represents a vast Improvement in scope and accuracy compared to MM. However, persisting to the present, MM approaches to molecular structure and shape prediction dominate across chemistry, despite its age and widely recognized and severe limitations. It survives because it can be applied with unmatched speed allowing it to scale over molecular size (towards proteins and microscale structures) and over library size (up to millions of molecules) whereas quantum mechanics has traditionally been orders of magnitude slower when applied to computational prediction. However, today there is no excuse, except for expediency and tradition, to continue to make predictions with MM with the advent of cloud computing and efficient alternative algorithms and tools to access quantum mechanics such as the faster density functional theory (DFT) and the ultra-fast semi-empirical methods.
ChemAlive has developed ConstruQt, to drive the shift away from MM to QM that is coming over the next few years. ConstruQt is high throughput quantum mechanics (QM), deployed on the cloud with full automation and is a first-of-its-kind tool for scaling molecular library design with the power of quantum mechanics. It dramatically increases molecular library accuracy in shape while adding energy-regime predictions for molecular structure such as tautomeric and diastereomeric selection and prioritization – both key to the design of active molecules.
A recent study of 750 conformationally diverse molecules has confirmed the extent to which classical force field approaches can be unreliable compared to higher-level data. The study further demonstrated the utility and accuracy of fast semi-empirical quantum mechanics as a paradigm shifting method in structural analysis. Thus, if molecular shape is being used in a molecular design workflow it is certainly generating false positives or filtering-off potential leads that would be captured by quantum mechanics. In addition, QM adds increasingly important scope towards reaction prediction and spectroscopic assignment.
Independently, we at ChemAlive have analyzed over 3 million molecules and over 200 million conformations using the Universal (UFF) and Merck Molecular (MMFF) force fields compared to the semi-empirical QM method PM6.
Our analysis reveals important differences in the global treatment of molecular shape by these methods. First of all, force fields over estimate conformer energy for strained systems thus creating false negatives and throwing out accessible conformers. The distribution in Figure 1 (a) shows a long tail for the UFF method reaching out to 20 kcal/mol.
Continue reading
This content available exclusively for BPT Mebmers
We use cookies to personalise content and to analyse our traffic.
You consent to our cookies if you continue to use our website. Read more details in our
cookies policy.