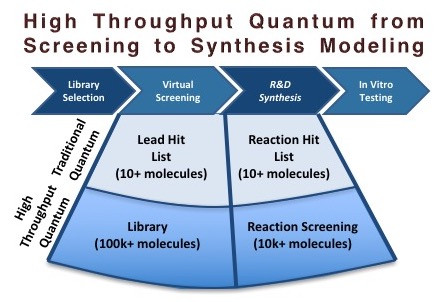
[White paper] High Throughput Quantum Chemistry for Drug Discovery - Towards Reaction Screening
In the domain of drug discovery, there can be a world of difference between a computer-generated hit compound, which is predicted to bind well to a drug target and what can be reliably synthesized at scale, or indeed synthesized at all. This discrepancy has been a lingering point of discord between the Discovery and R&D efforts in the chemical industry. Computer-aided drug design (CADD) has become an increasingly valuable tool by providing essential screening data and unique insight into drug action and mechanism, but it does not model the more complex world of chemical reactivity and synthetic chemistry.
Synthetic chemistry and computational chemistry traditionally overlap at the interface of physical organic chemistry (kinetics and mechanistic rational) and reaction design (often catalysis) where quantum mechanics is leveraged to provide quantitative predictions. There is a growing need for more quantum computations of reactions to control for this ‘obtainability’ factor extending to the optimization process as well, not just for R&D scale reactions but all the way through to production. Synthetic and process chemistry efforts carried out by medicinal chemists and chemical engineers carry an increasingly greater risk and cost to execute but suffer from not having their own computational CADD-like counterpart to achieve the same benefits that modeling is known to deliver at the beginning of the process.
In principle, theorists can now actually inform synthetic chemists how to synthesize that ‘impossible’ molecule, but this has come at a considerable cost in time and computing resources. Thus, in practice, the vast majority of computational work is performed on molecular structure and docking using molecular mechanics running on personal computers. This means that the interface between what could be a great drug and what lead compounds can actually be made is left with a technological gap where the analysis of the reaction transformations necessary to achieve these lead structures are left largely to literature searching or more high throughput data-driven methods like cheminformatics, and more recently, machine learning approaches.
State of Computation in Industry
Following increasing trends in digitization and computation, the chemical industry is preparing for an increasingly large user-base of chemical researchers and practitioners engaging with computations. There are over 1 million potential individual users working across thousands of innovation-driven companies. Roughly USD 19 billion p.a. will be spent on computation by 2020 within chemical R&D compared to about USD 9.5 billion currently.
As computation becomes more ubiquitous it will become an expected aspect of chemical workflow. This technology is becoming more central to the process of drug design as it provides metrics at the point in in the development and research cycle when ideas are transformed into real-world lead compounds.
Computation is still a niche market. The software sales market for traditional software packages in chemistry is This market is dominated by a number of high-end suites of programs. Accelrys and Schroedinger Inc. represent a large market share for general computational chemistry environments. In 2013, Accelrys had a revenue ca. 170 M USD, a growth of 3.7% and employed over 750 people with over 2000 clients. In 2008, Accelys was valued at 100 M USD. The company was later sold to Dassault Systems under the new label Biovia for 750 M USD in 2012. The market is clearly growing (5% per year). The purchase price for industrial agents for these software range from USD 2k - 50k. In the quantum area, Schroedinger is the most innovative and is recently moving into the cloud area with early elements of automation as well.
Continue reading
This content available exclusively for BPT Mebmers
We use cookies to personalise content and to analyse our traffic.
You consent to our cookies if you continue to use our website. Read more details in our
cookies policy.