Sponsored by Syntekabio
The State of A.I. Drug Discovery and Its Future: Small Molecules, Vaccines, and Antibodies
Artificial intelligence (A.I.) and big data in a synergistic integration have led to promising results in new drug development by drastically reducing cost and time. This trend in the biopharmaceutical market has emerged as a new industry standard as startups—each with distinctive core A.I. algorithm—fiercely compete to capture their global market share. Syntekabio, the first KOSDAQ-listed A.I. driven biotech company in South Korea, shares its view on the current and future trajectory of A.I. drug discovery and development in a series of exclusive articles for BiopharmaTrend.
Exploring A.I. Platform for New Drug Development
Companies that openly disclosed their A.I. drug development technologies so far include Schrödinger, Insilico Medicine and Atomwise.
Schrödinger has a 'free energy perturbation (FEP+)' platform that shows strength in precise binding energy calculation based on molecular dynamics, which is best used for Me-Too drug design in new drug development.
Insilico Medicine has Chemistry42, known as a generative A.I. drug design platform. This platform creates new compounds by using more than 30 generative models in parallel, and finally selects candidates in consideration of synthesis potential, novelty and diversity. An introduction of a robotic experiment also added to their strength.
An active A.I. driven drug development company since the beginning, Atomwise recently introduced A.I. based 'AtomNet PoseRanker (ANPR)' with improved performance.
Schrödinger, Insilico Medicine and Atomwise all offer innovative services for discovering new drug candidates that are superior to traditional methods. They can at once discover active and optimize lead substances because a large amount of lead substance derivatives is made when verification analysis is performed immediately after selecting an active substance on the A.I. new drug platform. This is also the reason why global pharmaceutical companies use their services despite the high cost. However, these services aren’t easily accessible for small and medium-sized pharmaceutical companies as well as new drug development research institutes and universities due to the cost.
Being amply aware of this service gap, Syntekabio launched STB CLOUD in late 2022, a suite of cloud-based proprietary platforms, to meet the unmet needs of these cost-conscious innovators seeking reliable yet affordable new drug candidate discovery services. Imagine screening 1 billion compounds that can be purchased with a single click anywhere, any time. STB CLOUD searches for the best pose in most targets through rapid recognition and deep learning. For 1,000 candidate substances at the final stage, it performs molecular dynamics simulations, and then finally selects 100 to 200 A.I.-hit candidates by free energy-based filtering out of 1,000 best poses.
What’s more is that STB CLOUD safely secures customer information, while the platform is completely automated and can be operated by anyone without professional training to quickly create a new drug development pipeline. Since the screening is performed based on commercially available compounds, it is also possible to discover new active substances with minimal synthesis, enabling the discovery of first-in-class compounds at an approximately four to five times lower than the average cost. This is a huge saving for most companies without compromising the quality of and timeline for new drug development.
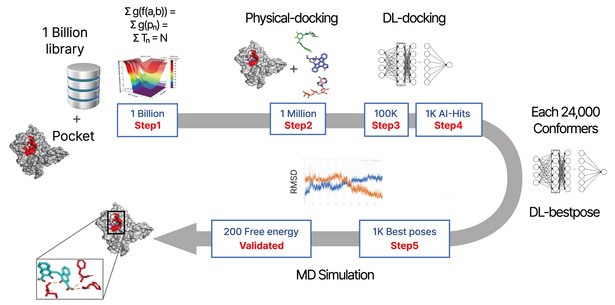
Figure 1. The flow diagram of DeepMatcher@Hit shows that compounds are first screened through three steps before being docked in a pocket of the target. Once the optimal pose is selected, the MD simulation is finally performed in a total of five steps.
Factors on Hit-discovery Success Rate of A.I. Drugs
The following three steps are essential for computer-based Hit-discovery: candidate compound search, optimal pose generation, and molecular dynamics verification. A.I. and advanced algorithms contribute greatly to the stage of searching for candidates and generating optimal poses. After searching for a candidate compound, an optimal pose can be generated so that the correct binding energy can be applied. Then the experimental verification is performed according to the ranking order of the calculated candidate compound.
It is fundamental to reduce errors in the experimental verifications to minimize the experiment cost, because the cost increases exponentially depending on the level of the verification amount, depending on 100, 1000, or 5000. When the third step—molecular dynamics—is applied to the best pose selected in the second step of discovering hit compounds, the optimal pose is created with structure closest to the experimental value. Several papers have cited greatly reduced errors even when ranking is calculated only by binding energy.
An internally conducted research also confirmed that adding a molecular dynamics verification step to top-notch A.I. and advanced algorithms more than doubled the ability to discover viable drug candidates. It is possible to derive active substances with excellent efficacy by excluding error substances, rather than simply finding active substances. Currently, among A.I. drug development companies such as Schrödinger, Insilico Medicine, Atomwise, and Syntekabio, only Syntekabio operates all three stages automatically.
Drug Efficacy Optimization (Auto-Lead-Effi-Opt)
Among the general drug optimization software available today, Syntekabio’s own software command line interpreter (CLI) called Gap2 stands out. A graphical user interface (GUI) in the traditional method makes it impossible to attach a large R-group (substituent) when making a derivative. Gap2, on the other hand, can do this efficiently in a supercomputer environment, directly attaching an R-group to a specific atom of the drug scaffold within the protein-drug binding pocket.
Syntekabio currently has about 20,000 of its own R-group libraries. According to the paper published by Schrödinger, the attempt to attach the R-group to the drug scaffold is carried out outside the protein and then brought into the protein’s pocket. Contrary to this, Syntekabio has developed structures with high activity among 20,000 R-groups directly from within the pocket. Hence, quantitative structure-activity relationship (QSAR) is no longer necessary as it automatically compares and selects in terms of binding energy. Therefore, Syntekabio's drug optimization is called Auto-Lead-Efficacy-Optimization. It is operated semi-automatically for now in the local environment, with an aim to be installed on STB CLOUD within the second quarter of 2023.
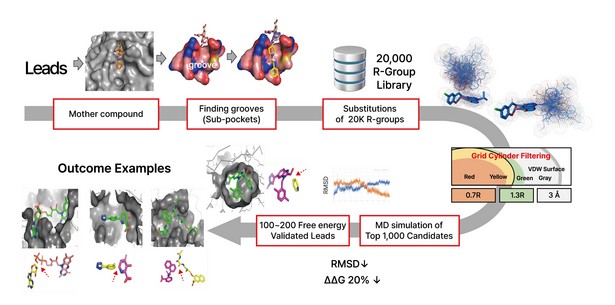
Figure2. Lead Optimization by DeepMatcher@Lead shows 20,000 R-groups are substituted and perturbed at the position of the mother compound, as the top 1,000 candidates are selected through grid cylinder filtering. Then 1,000 MD simulation is performed to complete the process.
DeepMatcher: an Innovative Platform for Discovering New Vaccines and Antibodies
Vaccines are a critical component of public health policy in eliminating serious infectious diseases, as seen in the recent Covid-19 pandemic. Making lifesaving drugs involves multiple forms and several processes in the manufacturing. Recent advances in vaccine technologies and platforms focusing on their mechanisms of action have opened a new pathway for the development of safe and efficacious formulations. In this context, Syntekabio's DeepMatcher, an A.I.-small molecule drug platform, has reached the stage of full commercialization.
DeepMatcher provides registry information for all residues in the pocket as output, thus acquiring analysis information of key residues involved in binding within the pocket possible when calculating the final binding energy. Thanks to this feature, it is easy to calculate registry information on interactions between new small molecule drugs (protein-ligand), new vaccine drugs (MHC-peptide), and new antibody drugs (antigen-antibody CDR).
A.I. based small molecule new drug discovery process using DeepMatcher involves three steps: candidate substance search, optimal pose generation, and molecular dynamics verification. The second and third steps here can be used together for calculating new vaccines and antibodies, shortening the development period on A.I. vaccine and antibody new drug platforms.
Complimentary to this technology, an A.I. vaccine drug platform NEO-ARS™ is in the verification stage, while Syntekabio develops AB-scan, an A.I. antibody drug platform. Neo-antigens discovered by NEO-ARS™ play a significant role in activating the immune system of cell therapy products from the market point of view. Cell therapy products have been growing gradually in the marketplace. It is greatly anticipated for the AB-scan platform to contribute to the new drug development once commercialized, especially in line with the growth of the markets for new antibody drugs and cell therapy.
At this time, DeepMatcher proceeds without interruption on a large number of CPU/GPU servers for three weeks, as STB CLOUD searches in three stages of operation for candidate compounds, optimal pose generation and molecular dynamics verification. In the first step, 1 million substances are selected from 1 billion compounds based on their physico-chemical properties and topology, and through physical docking with a fast docking algorithm, they enter deep learning docking and proceed to the final molecular dynamics verification.
The introduction of GPT, a hot topic these days, is being considered for implementation in the first screening stage to help improve the performance of DeepMatcher. Syntekabio is attempting proof of concept (POC) using GPT since its A.I. platform operates based on the registry information of all residues in the protein pocket, and these registries can be expressed as language models.
All stages of the drug discovery process are working in concert seamlessly to bring batter speed and performance, propelling DeepMatcher to rise above others at the epicenter of innovative IT technology. With continuous innovation, Syntekabio is poised to take the lead as a global hit-discovery and optimization CRO unmatched in the industry.