QUAISAR Computational Platform for Designing Targeted Protein Degraders
A key step in the targeted protein degradation (TPD) process is the formation of an induced proximity complex, where a degrader molecule recruits an E3 ligase to the protein of interest (POI), facilitating the transfer of ubiquitin to the POI and initiating the proteasomal degradation process.
Roivant Discovery is using their QUAISAR computational platform coupled with biophysical methods in the lab to address three critical aspects of induced proximity:
- formation of the ternary complex induced by a degrader molecule
- conformational heterogeneity of the ternary complex
- assessment of ubiquitination propensity via the full cullin-RING ligase (CRL) macromolecular assembly
These unique capabilities allow for the rational engineering of degrader molecules, atom by atom, using QUAISAR (QUantum, AI, and Structure-Activity Relationships), which t combines computational physics and artificial intelligence, with Roivant’s expertise in disease biology, chemistry, biophysics, proteomics, and translational informatics to accelerate the progression of drug discovery projects toward the clinic.
Their novel TPD approach integrates experimental biophysical data (hydrogen-deuterium exchange mass spectrometry; HDX-MS) with all-atom explicit solvent molecular dynamics (MD) simulations aided by enhanced sampling techniques to predict structural ensembles of ternary complexes. The result is an accurate dynamic model of the ternary structure at atomic resolution, as seen in this video. Their published validation work on the bromodomain of SMARCA2 with the E3 ligase VHL consistently and accurately reproduced known X-ray crystal structures. Interestingly, the simulations identified a structural ensemble of low-energy conformations of the ternary complex, suggesting that flexibility and dynamics of the ternary structure may be important for functional activity.
To further characterize the structural ensemble, the researchers at Roivant Discovery performed milliseconds of aggregate simulation time using Folding@home (the world’s largest supercomputer) to fully characterize the free energy surface around the various low-energy conformational states. These long-timescale simulations revealed that the crystal structure conformation exists within a broad low-energy basin. Encouragingly, he dynamic ensemble was found to be more consistent with solution-phase biophysical experimental data (HDX-MS and small-angle X-ray scattering; SAXS) than the crystal structure, giving further weight to the importance of solution dynamics. These ternary structures were then grafted onto the full CRL for additional enhanced sampling simulations, where it was found that differences in degradation efficiency could be explained by the proximity distribution of lysine residues on the POI relative to the E2-loaded ubiquitin (see Figure 3). Several of the top predicted ubiquitinated lysine residues have been validated prospectively through ubiquitin mapping proteomics experiments.
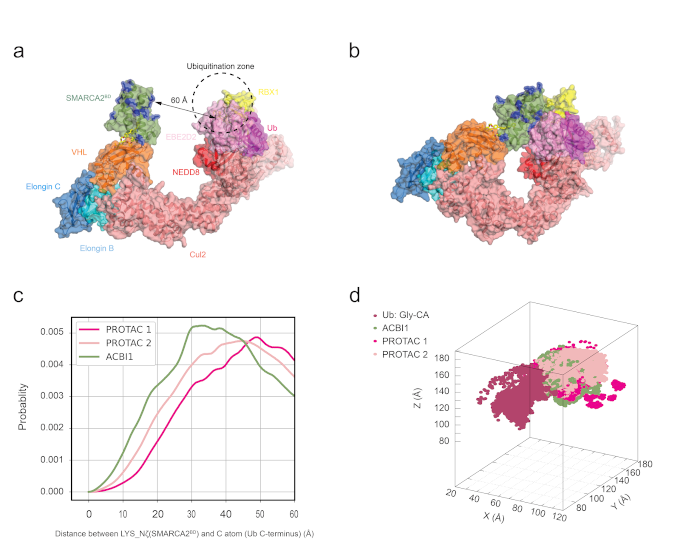
Figure 1: Lysine densities in the CRL-VHL ubiquitination zone from molecular dynamics simulations. a) VHL bound to SMARCA2BD and E2-ubiquitin in the open cullin-RING ligase conformation. b) Closed conformation of the CRL generated by QUAISAR simulations. c) SMARCA2BD lysine distance distribution to the C-terminal glycine of ubiquitin for the three different degrader molecules. d) Density of lysine residues in 3D space near the ubiquitination zone.
This simulation-driven approach using the QUAISAR platform is being actively applied to the Roivant Discovery TPD pipeline of oncology and inflammation targets, where chemical reaction transformations and machine learning are used to explore and triage thousands of design ideas while molecules are ultimately prioritized for synthesis using the aforementioned simulation approach. A more detailed description of the above work is available on the bioRxiv preprint server.