Some Drugs That Improve Price Competition Fall Through The Cracks Of FDA User Fees
Americans are increasingly concerned about rising drug prices. FDA user fees affect drug pricing competition, which is often overlooked. FDA user fees fund regulatory activities and encourage timely reviews and approvals, but some drugs that could improve price competition may fall through the cracks. Thus, smaller pharmaceutical companies and other generic drug or biosimilar manufacturers may face longer approval times or other regulatory hurdles, limiting their price competitiveness. FDA user fees and drug pricing competition will be discussed in this article.
Price competition and drug spending moderation require generic drugs. They account for 92% of prescriptions but only 16% of US invoice-level spending.
FDA approval of generic drugs' therapeutic equivalence allows price competition. That allows the pharmacy to automatically switch to the generic drug. This lets cheaper generics quickly overtake the more expensive brand. This article explores TE assessment delays, the FDA's regulations and practices for assigning TE codes, and the ways in which the FDA or Congress could address these issues.
Despite the competitive importance of therapeutic equivalence, the FDA does not assign TE codes at approval to all eligible drugs, leaving "money on the table" and disadvantaging American consumers.
This article explores TE assessment delays, the FDA's regulations and practices for assigning TE codes, and the ways in which the FDA or Congress could address these issues. Current legislative proposals that would set a 180-day deadline for FDA review of citizen petitions requesting TE codes would improve prescription drug market competition. FDA should include TE determinations at approval.
Therapeutic Equivalence Determination Governed By FDA Regulations And Practices
Therapeutically equivalent generics contain the same active drug ingredient in the same dosage, form, and route of administration as the reference brand name drug. They are bioequivalent to the branded product, meaning the active ingredient is absorbed at the same rate and extent. Changes to inactive ingredients, form, packaging, and shelf life are acceptable as long as the drug still has the same therapeutic effect.
The FDA gives drugs an A rating if they are therapeutically equivalent to other pharmaceutically equivalent products. Tablets and capsules get AB ratings. Drug formulations without bioequivalence issues are labeled AA, AM, AO, AP, or AT. AP is for pills, while AO is for oils. At approval, the ANDA assigns the TE code.
To obtain market authorization using the ANDA pathway, injectable drugs must contain, with some exceptions, the same inactive ingredients (Q1) and concentration (Q2) as the reference listed drug (branded product). Therapeutic equivalence allows for differences in inactive ingredients. Q1/Q2 sameness.
If branded products contain inactive ingredients, the Q1/Q2 sameness requirement can hinder entry. Generic manufacturers must avoid the ANDA pathway to avoid inactive ingredient patents. The FDA directs them to the 505(b)(2) pathway, where the Office of New Drugs reviews them instead of the Office of Generic Drugs. PDUFA user fees are higher, but turnaround times are faster than GDUFA. TE ratings exempt drugs from PDUFA product user fees.
Unlike with ANDAs, the FDA does not automatically assign TE codes to 505(b)(2) applicants. Generic companies must file citizen petitions after FDA approval.
Evaluation of the FDA's Processing of Requests for Therapeutic Equivalence Determinations Under Section 505(B)(2)
We reviewed citizen petitions for A-ratings to evaluate the 505(b)(2) pathway for these drugs. The number of citizen petitions was 22. These petitions represent 28 injectable drugs. 25 want AP ratings, two AB ratings, and one AO rating. 20 of the 28 are intravenous-only, which should automatically qualify them for a biowaiver and an AP rating if the drug is pharmaceutically equivalent. The remaining eight drugs are injected subcutaneously or intramuscularly. These routes may need more data for an A rating.
FDA has delayed 505(b)(2) citizen petitions, as shown below. Xellia's voriconazole for injection was the only FDA-approved A-rating request from 22 petitioners. That petition took 654 days. One petition was denied by the FDA because the reference brand drug was no longer manufactured. Four petitioners withdrew, one by discontinuing drug marketing. Most others have waited two or three years.
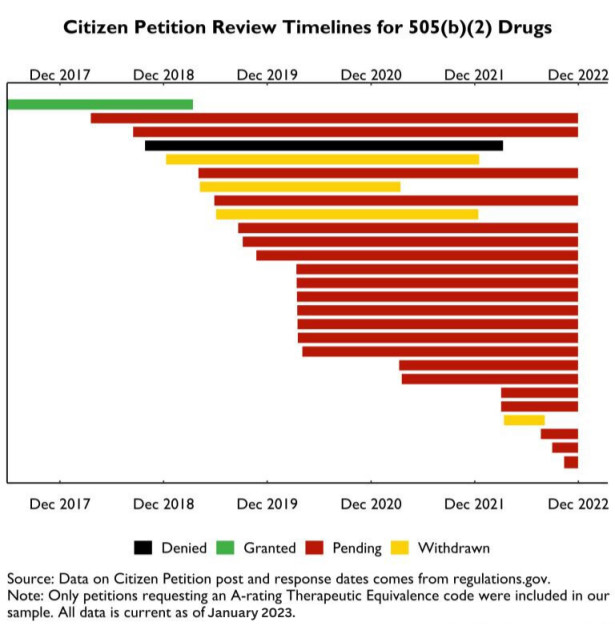
FDA delays in processing citizen petitions have failed to promote competition, raising consumer and purchaser costs. A Medicare payment change highlights the cost of these delays. CMS issued guidance in November 2022 to issue separate reimbursement codes for unrated generic 505(b)(2) drugs. CMS included those drugs in the same HCPCS code as the branded product, lowering brand reimbursement and encouraging providers to switch to higher provider margin 505(b)(2) alternatives.
The CMS guidance, which legally aligns the multisource drug determination with FDA's TE determination, means that these companies lose the benefits of competition unless they get A-ratings, making FDA's TE rating process more important.
Methods for Streamlining the Approval of Requests for a 505(B)(2) Therapeutic Equivalence Determination
FDA told petitioners that 505(b)(2) TE petitions are not a priority. “FDA has been unable to reach a decision on your petition due to the need to address other Agency priorities,” a September 2020 follow-up letter stated. Given the Agency's many demands, we will respond to your petition as soon as possible.” Petition pending.
User fee process improvements, Congressional mandates, and executive branch engagement are three ways to speed up TE petition consideration.
Streamlining user fees
User fees solve review process inefficiencies and resource constraints. FDA and industry identify inefficient processes, agree on process changes, funding, and performance commitments.
Unfortunately, 505(b)(2) TE rating requests are in a gray area between the two main user fee domains. Section 505(b)(2) TE applicants are generic companies using GDUFA processes. PDUFA funds the 505(b)(2) pathway, which TE petitioners rarely negotiate. Any additional TE review work in the 505(b)(2) process, no matter how small, could compromise the FDA's PDUFA timelines and its commitments to stakeholders. PDUFA changes are unlikely to solve this issue.
GDUFA may allow a by-request TE rating review. Fees would fund the FDA and set performance goals. GDUFA TE and PDUFA 505(b)(2) reviews can be aligned, but it's difficult. Since user fees won't change again until 2027, this policy direction must be long-term.
Congressional mandate
The issue is one of resource and priority, not mandate. FDA prefers the citizen petition process in the current user fee environment, but Congress can order FDA to speed up these reviews. Congress may pass a year-end package rider that sets 180-day timelines for reviewing citizen petitions.
A timeline would help the FDA. We recommend more appropriations to address resource constraints. Given the low volume and simplicity of these TE requests—remember that most of these drugs are intravenous only and should qualify for biowaivers, which automatically determine bioequivalence—those appropriations would likely be modest. Despite funding and a congressional mandate, we worry that priorities may remain elsewhere.
Executive involvement
Even with a 180-day Citizen Petition review timeline, post-approval TE code determinations via Citizen Petitions delay a small number of 505(b)(2)s. Delays violate President Biden's July 2021 Executive Order on Competition, which encouraged drug pricing policy.
The White House can encourage the FDA to improve its TE ratings by integrating them into future 505(b)(2) approvals instead of requiring citizen petitions. FDA could request funds from the President's Budget to address the Citizen Petitions backlog and incorporate by-request determinations into the approval process, which the Administration should support given President Biden's executive order.
References:
- The use of medicines in the U.S. 2022. IQVIA. (n.d.). Retrieved from https://www.iqvia.com/insights/the-iqvia-institute/reports/the-use-of-medicines-in-the-us-2022
- Center for Drug Evaluation and Research. (n.d.). Preface to the orange book. U.S. Food and Drug Administration. Retrieved from https://www.fda.gov/drugs/development-approval-process-drugs/orange-book-preface
- Center for Drug Evaluation and Research. (n.d.). Preface to the orange book. U.S. Food and Drug Administration. Retrieved from https://www.fda.gov/drugs/development-approval-process-drugs/orange-book-preface
- Center for Drug Evaluation and Research. (n.d.). Preface to the orange book. U.S. Food and Drug Administration. Retrieved from https://www.fda.gov/drugs/development-approval-process-drugs/orange-book-preface
- Navigating Q1/Q2 for complex generics - association for accessible ... (n.d.). Retrieved from https://accessiblemeds.org/sites/default/files/2019-11/Xiaohui_%28Jeff%29_Jiang_GRxBiosims2019.pdf
- Center for Drug Evaluation and Research. (n.d.). Determining whether to submit an anda or a 505(b)(2) application. U.S. Food and Drug Administration. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/determining-whether-submit-anda-or-505b2-application
- Prescription drug user fee act waivers, reductions, and refunds for ... (n.d.). Retrieved from https://www.fda.gov/media/131797/download
- Center for Drug Evaluation and Research. (n.d.). Preface to the orange book. U.S. Food and Drug Administration. Retrieved from https://www.fda.gov/drugs/development-approval-process-drugs/orange-book-preface
- FDA citizen petitions requesting a rating for 505(b)(2) drugs. (n.d.). Retrieved from https://www.brookings.edu/wp-content/uploads/2022/12/FDA-Citizen-Petitions-requesting-a-rating-for-505b2-drugs.pdf
- Regulations.gov. (n.d.). Retrieved from https://www.regulations.gov/document/FDA-2017-P-3329-0004
- Regulations.gov. (n.d.). Retrieved from https://www.regulations.gov/document/FDA-2020-P-1065-0004
- The United States Government. (2021, July 9). Executive order on promoting competition in the American economy. The White House. Retrieved from https://www.whitehouse.gov/briefing-room/presidential-actions/2021/07/09/executive-order-on-promoting-competition-in-the-american-economy/