Unveiling Hidden Drug Targets
Scientists estimated that the human genome encodes above 20,000 different proteins in our body. However, available public databases contain records of known ligands for only about 10% of all proteins. Expanding the map of “druggable” proteins increases the chances of running into novel small molecule drugs.
A recent study, published in Cell, opens doors for unveiling novel drug targets and even reconsidering some of the known protein targets, previously tagged “undruggable”.
The team led by scientists at The Scripps Research Institute (TSRI) has developed a chemical proteomic method to globally map reversible interactions between small molecule fragments and proteins directly in human cells. The new method enables researchers to quickly find small molecules (ligands) binding to hundreds of thousands of proteins in their native cellular environment with the exact identification of the proteins involved.
The newly introduced method involves, first, the development of a small but structurally diverse library of candidate ligand molecules - fragments. Then, each candidate ligand is decorated with a constant tag bearing an alkyne and photoactivatable diazirine group. Thus modified ligands are called Fully Functionalized Fragments (FFF).
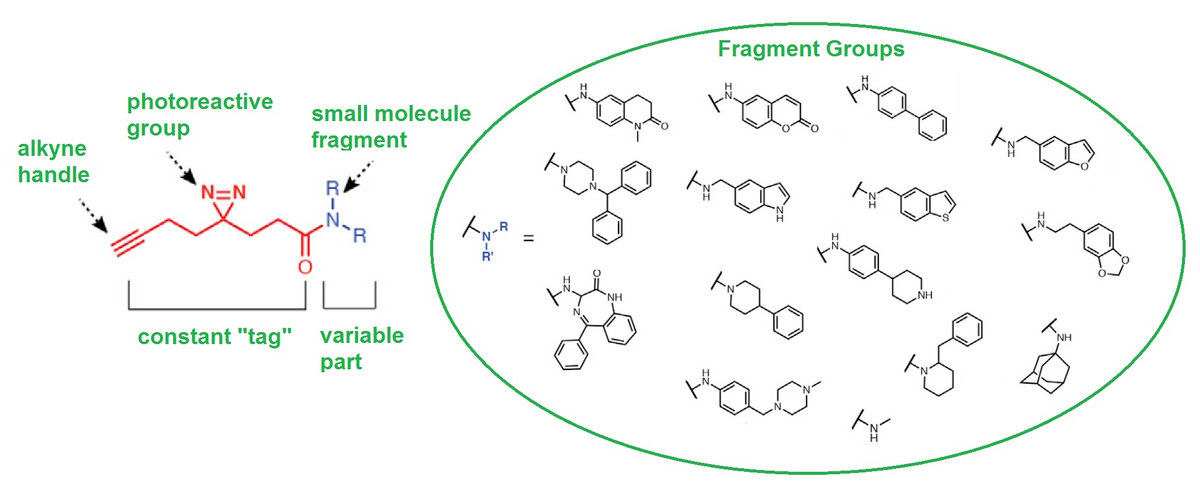
When the modified ligand binds with moderate affinity to a protein, a brief exposure to UV light activates the former making it stick permanently by forming a covalent bond.
Next, scientists use the alkyne group, a molecular handle, to grab and isolate the newly formed ligand-protein pair for analysis.
The authors also demonstrated that using the new method they can generate selective ligands modulating the functions of proteins in cells. Several types of biological targets were successfully investigated, including enzymes (PTGR2), transporters (SLC25A20), and poorly characterized transmembrane proteins (PGRMC2), lacking previously known selective ligands.
Applying initially a small library of just 11 drug-like candidate ligands to human cells, the researchers managed to identify more than 2,000 distinct proteins that had bound to one or more of the ligands.
Among those, there were proteins that previously had been considered “un-targetable with drugs, such as transcription factors.
Finally, the team from TSRI, together with their colleagues at Bristol-Myers Squibb, demonstrated the efficacy of the new method in a more complex use case. They created a library of several hundred candidate ligands and then applied them in a functional screen to reveal hits that could promote the maturation of fat cells - adipocytes. This process can alleviate the insulin resistance, related to type 2 diabetes.
With this new discovery method, the researchers identified not only a ligand that strongly promotes adipocyte maturation but also its binding partner - a poorly explored PGRMC2 protein. Thus, researchers found a new “druggable” pathway in a process, which has been extensively studied before.
The new method can become a powerful tool not only for identifying novel drug candidates but also for better understanding the very biology behind the scene. Conducting functional screens with this new approach, it is possible to immediately “map” the proteins or other molecules, responsible for the occurred biological effect in the cell -- without any prior knowledge of protein targets involved.
Topics: Tools & Methods