MIT and Recursion Launch Boltz-2, Open-Source AI Foundation Model for Structure and Binding Affinity Prediction
Researchers at MIT’s Jameel Clinic and CSAIL, in collaboration with Recursion and leveraging NVIDIA’s BioHive-2 supercomputer, have released Boltz-2, an open-source biomolecular foundation model designed to simultaneously predict 3D protein structures and their binding affinities.
Boltz-2 extends beyond earlier innovations such as AlphaFold3 and its own predecessor Boltz-1 (released last year) by integrating affinity predictions with dynamic structural modeling—achieving near-physics-based (FEP) accuracy up to 1000 times faster than traditional computational methods.
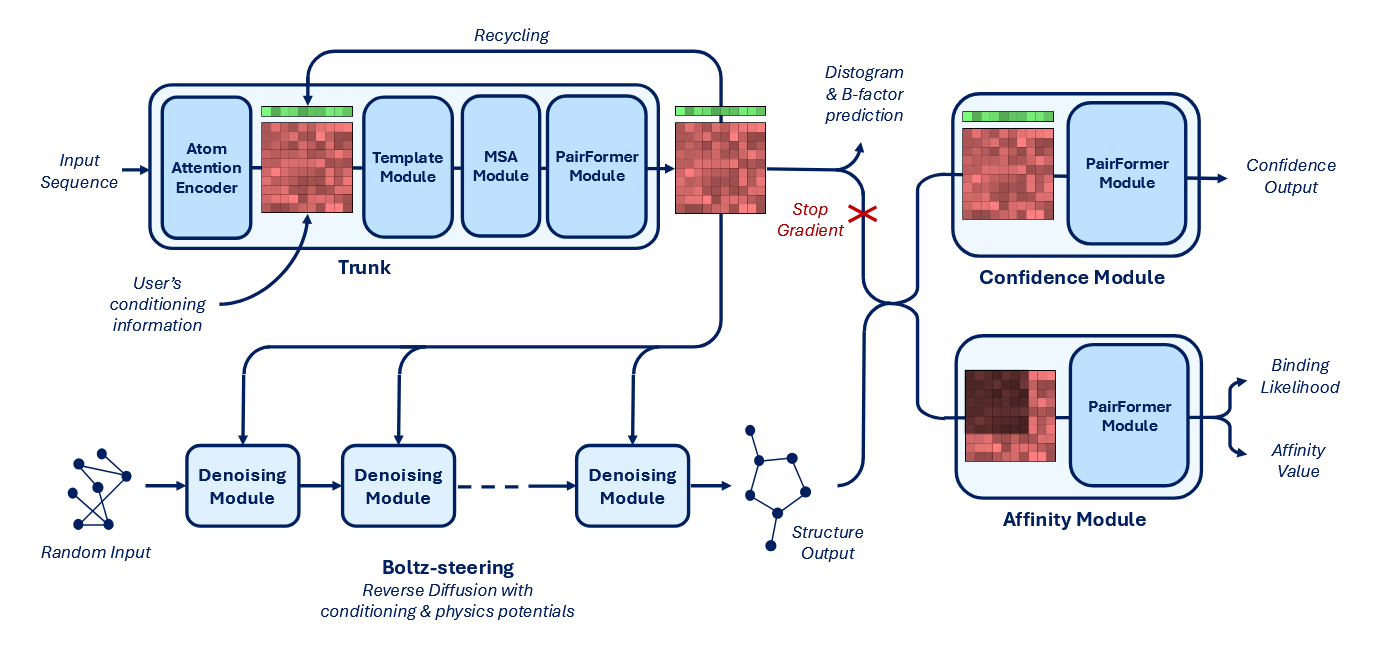
From paper: Boltz-2 model architecture diagram.
Boltz-2 was trained on a large dataset combining ~5 million binding affinity measurements, molecular dynamics simulations, and distillation data. It builds on Boltz-1 by adding a new affinity prediction module, GPU-level optimizations, and a technique called Boltz-Steering, which improves the physical realism of predicted structures. The model also supports user control through contact constraints, template guidance, and method-specific alignment.
See also: Foundation Models: Startups, Industry Updates and the Nobel Prize
Unlike models that treat structure and affinity as separate tasks, Boltz-2 predicts both jointly. It matches the accuracy of free energy perturbation (FEP) methods—physics-based simulations commonly used to estimate binding strength—while running over 1000× faster. In CASP16, it ranked first on binding affinity prediction across 140 complexes. It also performs well on DNA–protein, RNA, and antibody–antigen systems, and captures local dynamics more effectively than Boltz-1 when conditioned on MD data.
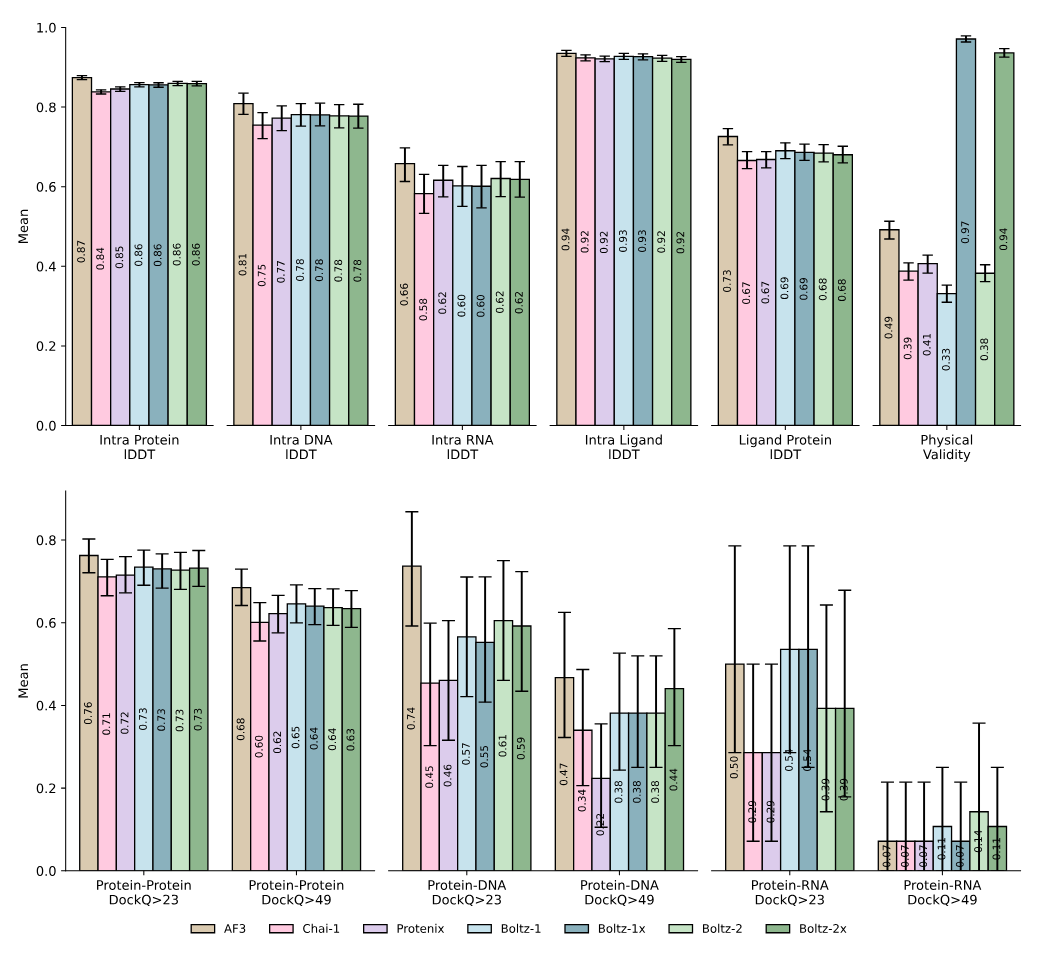
From paper: Evaluation of the performance of Boltz-2 against existing co-folding models on a diverse set of unseen complexes. Error bars indicate 95% confidence intervals.
Recursion Co-founder and CEO Chris Gibson contextualized Boltz-2 within a broader vision previously shared at JPM in January, in which he outlined Recursion’s ambition to model biology comprehensively, from atomic details to systemic pathways. On the significance of Boltz-2’s release, Gibson stated:
"Most importantly, Boltz-2 is fully open-source for all academic and commercial uses. With this release, we are entering an era where structural biology predictions will no longer be a differentiator nor a barrier—they are broadly accessible to all. This levels the playing field, opening up new opportunities for small startups and academic labs to be competitive with larger institutions as the field builds on open-source methods like Boltz-2.
It also shifts where value is created and differentiated in the drug discovery process. The focus turns to how well you can integrate this ‘protein layer’ with other multiomics data layers to get a complete view of biological systems; how well you can create an iterative cycle of dry-lab predictions and experimental wet-lab validation; how well you can build modular, adaptive, end-to-end platforms that tie everything together. That’s the long game we've been playing at Recursion since day one, and Boltz-2 accelerates that vision for the entire field."
The development of Boltz-2 was led by MIT professors Regina Barzilay and Tommi Jaakkola, with key contributions from researchers Gabriele Corso, Saro Passaro, and Jeremy Wohlwend, alongside a joint team from MIT and Recursion. Corso told Endpoints News he expects AI models of this class to reach experimental-level accuracy in binding affinity prediction within one to two years.
Boltz-2’s full training pipeline, model weights, and code are publicly available under an MIT license, accessible at boltz.bio/boltz2.