Arc Institute Releases its First Virtual Cell Model
Arc Institute has launched its first virtual cell model, State, designed to predict gene expression changes in response to chemical, genetic, or cytokine perturbations across diverse cell types. The model is publicly released for noncommercial use and trained on observational data from nearly 170 million cells and perturbational data from over 100 million cells across 70 human cell lines, making it the largest single-cell perturbation model to date.
State consists of two main modules:
- State Embedding (SE), which maps transcriptomes to a smooth vector space for robust comparison across cell types
- State Transition (ST), a bidirectional transformer that models how perturbations shift a cell’s gene expression profile in that latent space
The model outperformed existing computational baselines in benchmark tests. On the Tahoe-100M dataset (open-sourced by Vevo/Tahoe Therapeutics in February this year), State improved discrimination of perturbation effects by 50% and doubled the accuracy of identifying true differentially expressed genes compared to prior models. According to Arc, it is also the first model in this domain to consistently exceed the performance of simple linear methods.
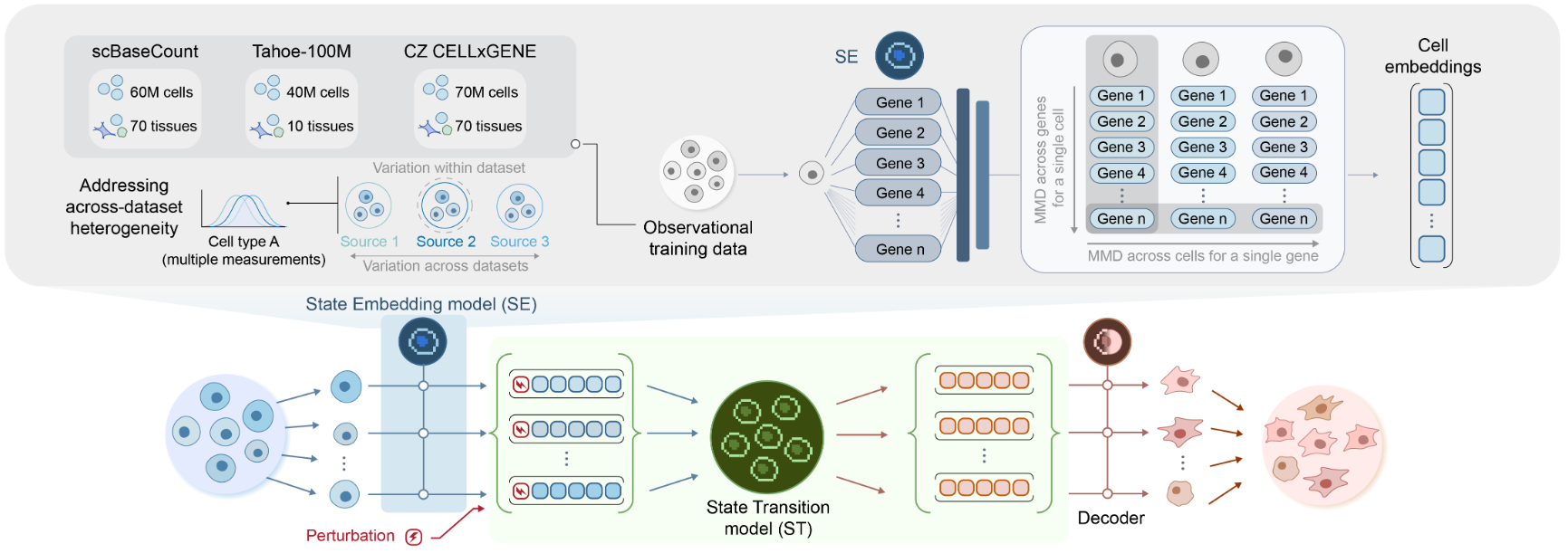
From manuscript, p.12, Figure 3: Figure 3: "State embeddings enhance zero-shot perturbation effect prediction across datasets, experiments, and modalities. (A) The State Embedding model (SE) learns rich, generalizable representations of transcriptomic information across diverse datasets. Given a control(unperturbed) cell population, SE computes cell embeddings. ST then predicts how those embeddings shift in response to a specified perturbation, effectively modeling the distributional effect of the perturbation in latent space. Finally, a learnt decoder maps the predicted embeddings back into gene expression space."
A central challenge in predictive cellular modeling is disentangling correlation from causation. To address this, Arc emphasizes large-scale perturbation datasets (including CRISPR-based gene knockouts and chemical interventions) as a more reliable signal of causal effects compared to purely observational RNA-seq data. The model was trained using data from Tahoe-100M, Parse-PMBC, and Replogle-Nadig, covering hundreds of perturbations across diverse contexts.
To support future model assessment, Arc has also introduced a "Cell_Eval" benchmarking framework tailored for virtual cell models. Unlike traditional RNA expression metrics, Cell_Eval includes biologically relevant metrics for perturbation strength estimation and differential expression prediction, designed to support transparency and standardization in future model development.
Underlying the project is Arc’s in-house infrastructure with scBaseCount (link to Arc's technical report), an AI system for harmonizing large-scale single-cell datasets across labs and technologies. Combined with Arc's vertically integrated experimental pipelines, scBaseCount enables fast iteration on data quality, scale, and modeling approaches.
Arc frames State as the first in a planned series of increasingly accurate models, aiming to enable large-scale in silico hypothesis generation, analogous to how protein folding models like AlphaFold have impacted drug design workflows. State is available via preprint and GitHub for researchers exploring virtual screening and mechanistic interrogation of cellular responses.
This February, Arc Institute and collaborators also released Evo 2, a foundation model for genomic research trained on 9.3 trillion DNA base pairs from over 128,000 genomes across all domains of life. Scaled up to 40 billion parameters, Evo 2 can process sequences up to 1 million base pairs, enabling prediction of variant effects, identification of regulatory elements, and full genome generation. It is publicly available via NVIDIA BioNeMo, with open access to model weights, training data, and inference tools.
Topic: AI in Bio