How AI Is Transforming Medicinal Chemistry: From Discovery to Manufacturing
In medicinal chemistry, the “make” stage of the design–make–test–analyze (DMTA) cycle is often where good ideas go to die. Synthetic bottlenecks, expensive building blocks, low-yield reactions, and complications during scale-up for manufacturing can turn promising drug candidates into abandoned projects.
Over the past decade, machine learning has emerged as the field’s most touted problem-solver, capable, in theory, of planning synthetic routes faster and in some cases, even on par with the most seasoned chemist. From transformer-based reaction-prediction models (e.g., the Molecular Transformer) to open-source academic initiatives like ASKCOS, and commercial platforms such as Molecule.one, Spaya, Reaxys Predictive Retrosynthesis, SYNTHIA®, SciFinder, and ChemAIRS, retrosynthetic planning has become an AI playground.
These tools can generate full multistep routes in minutes, rank analog series by their synthetic accessibility, flag high-risk transformations, and in some cases even propose reaction conditions. In practice, they are being used in pharma and biotech to support hit-to-lead optimization, parallel analog synthesis, and process-route design—often bridging the gap between early medicinal chemistry and later manufacturing considerations.
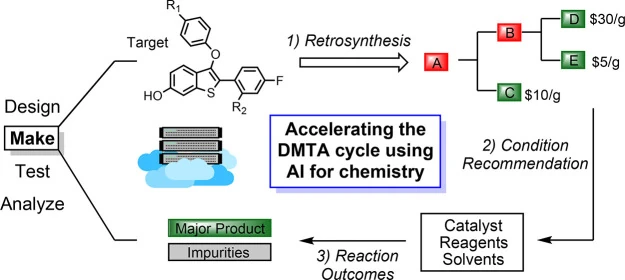
Image source: Struble TJ, Alvarez JC, Brown SP, et al. Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis. J Med Chem. 2020;63(16):8667-8682. doi:10.1021/acs.jmedchem.9b02120. Copyright: American Chemical Society (Licence: CC-BY)
The following use cases capture how AI is reshaping the synthesis side of medicinal chemistry today, drawing from both the lab bench and the server rack.
1. Retrosynthesis as a Baseline Capability
Retrosynthetic planning, i.e., systematically breaking a target molecule down into purchasable starting materials, is the cornerstone of AI-assisted synthesis planning.
In its simplest form, retrosynthesis is a search problem: starting from the target, identify a sequence of known reactions that ends with available building blocks. But in practice, it’s more than just finding a sequence of possible chemical reactions -- it’s about finding routes that balance feasibility, cost, yield potential, and, increasingly, supply-chain resilience.
Today’s AI retrosynthesis tools draw from a mix of methodologies, though their origins often shape how they work in practice. Platforms such as SciFinder’s retrosynthesis, Merck’s SYNTHIA®, and InfoChem’s ICSynth have long histories with extensive libraries of human-curated reaction templates, giving them strong coverage in specialized chemistries and a clear link to literature precedent.
Others, including Molecule.one, Spaya by Iktos, and the open-source ASKCOS, grew out of efforts to learn reactivity patterns directly from large reaction corpora, enabling rapid adaptation to new transformations and emerging chemical spaces.
Some newer entrants, like Chemical.AI’s ChemAIRS, weave together both traditions from the ground up, combining large-scale reaction datasets with algorithmic search that considers building-block availability, step efficiency, and scalability, thereby connecting early medicinal chemistry design with the practical demands of process development.
One academic example comes from the Machine Learning for Pharmaceutical Discovery and Synthesis (MLPDS) consortium, where ASKCOS has proposed viable routes to targets absent from its training data, including branebrutinib, using combinations of standard transformations like C–N couplings and heterocycle formations.
In the commercial sphere, Chemical.AI has documented a number of retrosynthesis case studies on its technical blog, illustrating how such tools can be applied to real-world medicinal chemistry projects.
One example of how AI is aiding drug synthesis is the ChemAIRS-proposed retrosynthesis of Elironrasib (RMC-6291), a next-generation KRAS-G12C(ON) inhibitor developed by Revolution Medicines. Unlike earlier compounds that target the inactive, GDP-bound form of KRAS, Elironrasib selectively engages the active, GTP-bound state by forming a tri-complex with cyclophilin A, offering enhanced potency and selectivity.
To suggest the synthesis of this complex macrocyclic molecule, ChemAIRS generated a detailed 25-step synthetic route that closely mirrors the patented strategy while introducing meaningful efficiencies.
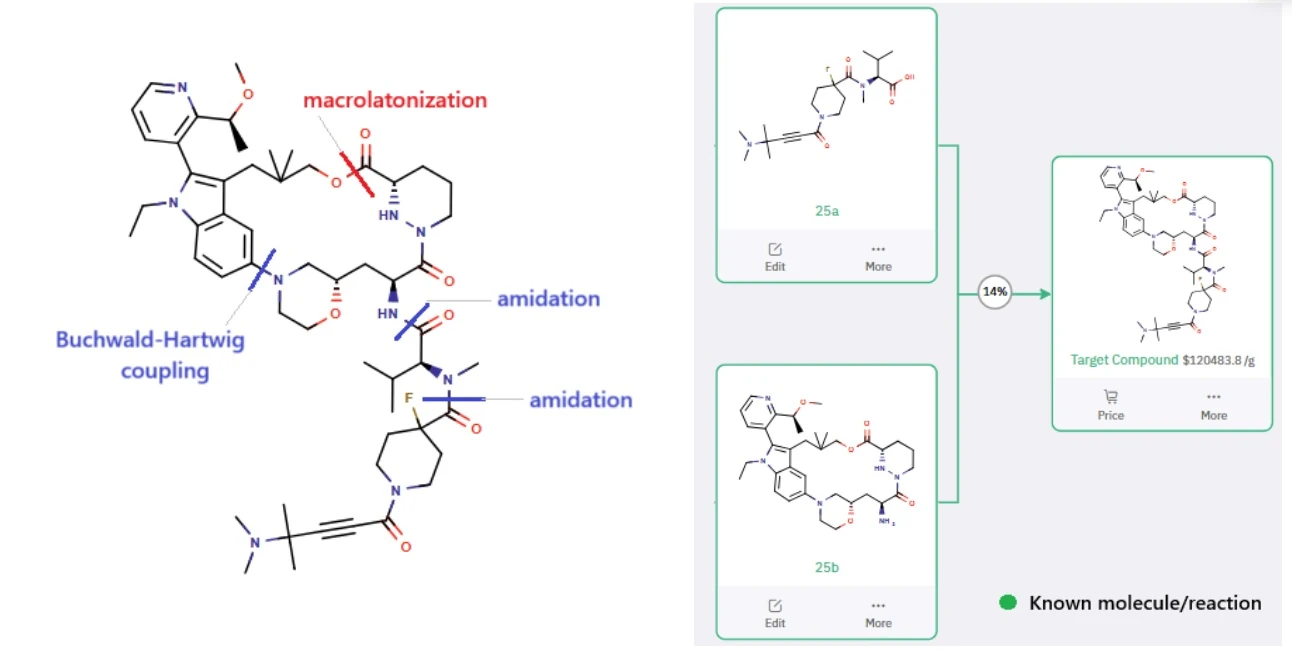
Image source: “ChemAIRS-Driven Retrosynthesis of Elironrasib (RMC-6291): A Next-Gen KRAS-G12C(ON) Inhibitor_EP20” – a case study by Chemical.AI (with permission)
By breaking down the target into two key intermediates, ChemAIRS enabled a modular approach and proposed streamlined synthetic pathways using commercially available building blocks, including a novel, cost-effective three-step synthesis of a key component that reduced reliance on expensive precursors.
Beyond route design, the platform also flagged potential side reactions, showcasing its value not only in accelerating synthesis planning but also in mitigating downstream development risks—an example of how AI is reshaping the landscape of medicinal chemistry.
In all cases, the technology acts as a co-pilot, helping in accelerating the exploration of possible strategies. When a retrosynthesis engine can deliver multiple viable options, complete with literature precedent, starting material availability, and realistic step counts, it changes the starting point of synthesis planning from “Can we make it?” to “Which of these do we make first?”
2. Route Scouting and Synthetic Accessibility at Scale
Retrosynthesis, breaking a target down to starting materials, can tell you whether a molecule is theoretically makeable. Route scouting goes a step further: it compares multiple viable synthetic pathways and weighs them against practical constraints, including cost, robustness, supply-chain security, and, crucially, scalability from milligram medicinal chemistry batches to multi-kilogram process runs.
In other words, it’s about choosing not just a chemically valid route, but one that will survive the transition to manufacturing. The smart route scouting starts in the early discovery stage, rather than waiting till preclinical and clinical stages. This helps avoid “turbulence” and unnecessary costs and project delays when scaling up synthesis.
AI is transforming this comparative stage by making it possible to generate and evaluate multiple synthetic options quickly. Within GSK, for example, ASKCOS was used to produce alternative routes for a large set of candidate molecules. By expanding its building-block database from 138,000 to more than 8 million commercially available compounds, route-finding success increased from 54% to 67%, opening access to new chemical space that would have been considered inaccessible using a narrower set of inputs.
In another illustrative example, Chemical.AI used ChemAIRS to scout routes for challenging macrocyclic 3CLpro inhibitors targeting the coronavirus main protease. The platform proposed an alternative macrocyclization strategy, a Pd-catalyzed intramolecular Buchwald–Hartwig C–N coupling in place of the patented macrolactamization, alongside fragment synthesis routes optimized for cost and scalability. The result was a set of viable options balancing novel disconnections with supply-chain practicality.
3. Forward Prediction — Avoiding Synthetic Dead Ends
While retrosynthesis works backward from a target molecule and route scouting compares full synthetic pathways, forward prediction operates at the reaction level. It anticipates how a proposed transformation will behave in practice, identifying the main product, possible side products, and even condition sensitivities, before any bench work begins. This capability acts as a safety net, flagging potential pitfalls such as regioisomer mixtures that complicate purification, functional-group incompatibilities that could stall a key step, or unexpected byproducts that threaten overall yield.
At Pfizer and the University of Cambridge, retraining sequence-to-sequence models on proprietary electronic lab notebook (ELN) data markedly improved the accuracy of reaction outcome predictions, making them more reflective of real-world in-house chemistry.
Extending the concept further upstream, Chemical.AI combines molecular design with route feasibility checks. It allows chemists to create virtual molecular libraries where each candidate is evaluated for synthetic accessibility from the outset, filtering out structures likely to be unstable, reactive, or impractical. The result is a set of novel designs that are not only chemically interesting but immediately actionable in the lab.
From Milligrams to Metric Tons: Where AI Still Has to Prove Itself
In medicinal chemistry, AI has become the lab’s tireless ideator—sketching synthetic routes in seconds, ranking analog libraries by synthetic accessibility, and even predicting when a Buchwald–Hartwig coupling might give a messy regioisomer mix. But crossing from the gram scale of discovery chemistry to the multi-kilogram scale of process manufacturing is not a matter of running the same playbook faster.
On scale, step count is a poor proxy for viability. Process mass intensity (PMI), solvent usage, impurity rejection, crystallization yield, and raw material availability drive the economics and regulatory acceptance of a route. An elegant five-step synthesis that generates hard-to-purge organometallic residues or relies on a thermally unstable intermediate can be dead on arrival at the plant. Predicting these pitfalls requires AI models that go beyond static reaction templates to incorporate kinetics, thermodynamics, and phase-behavior modeling. For example, a robust scale-up-oriented planner should be able to simulate heat profiles to flag exotherms, estimate solvent–solute partitioning to predict liquid–liquid extraction efficiency, and model polymorph formation in mixed-solvent crystallizations.
This is where the future points toward integrated digital twins for process development—systems that combine mechanistic simulations with continuously retrained ML models, updating impurity-formation pathways, solvent effects, and crystallization behavior in real time from plant data. Instead of a single yield number, such a twin could output PMI, predicted impurity carryover, energy demand, and regulatory classification of intermediates.
Recent academic research projects hint at what this next stage could look like. Multi-agent systems such as the ChemAgents framework demonstrate how specialized AI components, literature parsers, experiment designers, computational modelers, and robotic operators, can work in concert under a coordinating layer, with each experiment’s results automatically refining the next round of planning.
While still early days, commercially available tools are edging toward this vision, too. Platforms like ChemAIRS are already integrating retrosynthesis, forward prediction, and route evaluation in ways that account for building-block availability, supply-chain constraints, and potential scale-up challenges, bringing the concept of an AI-driven, closed-loop process architect closer to reality.
Topic: AI in Bio