AI Becomes a Drop-in Replacement for Docking
The field of AI ligand pose prediction in the protein binding pockets (aka “AI docking”) witnessed unprecedented development in the last year. Several research groups and companies have presented their recent achievements that compete with each other on the dedicated docking benchmarks. The most widely used is the PoseBusters dataset, which is specially tailored to challenge the AI docking technique in terms of the chemical and conformational correctness of predicted binding poses.
One of the leading players in this emerging niche is London-based startup company Receptor.AI, which recently presented its flagship AI docking model, ArtiDock v1.5 that demonstrated impressive performance and speed on the PoseBusters dataset [link]. A few days ago, the company made another big announcement about ArtiDock v2.0 – an even more advanced version of their technique with stellar benchmark metrics. This time the announcement is backed up by a preprint, which discloses important details about the model architecture, training, and tuning.
Explaining their technological choice Receptor.AI notes that the first generation of AI docking techniques used simple and lightweight model architectures and demonstrated results that were inferior to conventional docking. This issue was mitigated to some extent in the second generation of AI docking models by using much heavier architectures. The boost of accuracy, however, came at the expense of very complex architectures, large model sizes, and, as a result, very slow training and inference.
ArtiDock exploits the opposite approach by providing a deliberately lightweight and fast model architecture, which is trained on a much larger amount of augmented data from several sources. This results in better prediction accuracy without compromising the inference speed.
ArtiDock is based on a proprietary model architecture inspired by lightweight trigonometry-aware neural networks. The training set includes the PDBbind database (v. 2020), a collection of biomolecular complexes from the PDB with experimentally measured binding affinity data, and the Binding MOAD database (v. 2020), which provides a subset of the PDB, containing all high-quality ligand-protein complexes irrespective of the availability of the binding affinity data.
In addition to this two sources of augmented data were used:
- The PocketCFDM approach [] algorithm augments the training set of the protein-ligand complexes with artificial data, which mimics real protein binding pockets in terms of statistical distributions of the non-bond interactions. This method allows for generating artificial binding pockets for arbitrary small molecule conformers and was previously proven to produce high-quality augmentation data.
- The results of massive MD simulations of about ~17,000 protein-ligand complexes extracted from PDBbind v.2020. The ligands were processed and subjected to ab initio quantum simulations, while the protein structures were treated using the proprietary structure preparation module of the Receptor.AI platform. All complexes were simulated for a fixed amount of wall time, which resulted in system size-dependent simulation times with a median of ~30 ns. The trajectories were clustered using proprietary analysis routines, resulting in ensembles of representative conformations that were randomized and used for the training set augmentation.
ArtiDock predicts the matrix of the protein-ligand interactions, which is then converted into the 3D pose of the ligand utilizing a custom distance matrix-to-pose algorithm, which is based on the differential evolution (DE) technique with additionally enforced penalization of steric clashes. The latter is very important to avoid steric clashes and multiple other quality issues with predicted binding poses, which are common for the majority of the other AI docking techniques.
The authors also compared several versions of ArtiDock that differ by the training dataset and the distance matrix-to-pose algorithm used, which allows us to estimate the roles of these factors in the model’s performance.
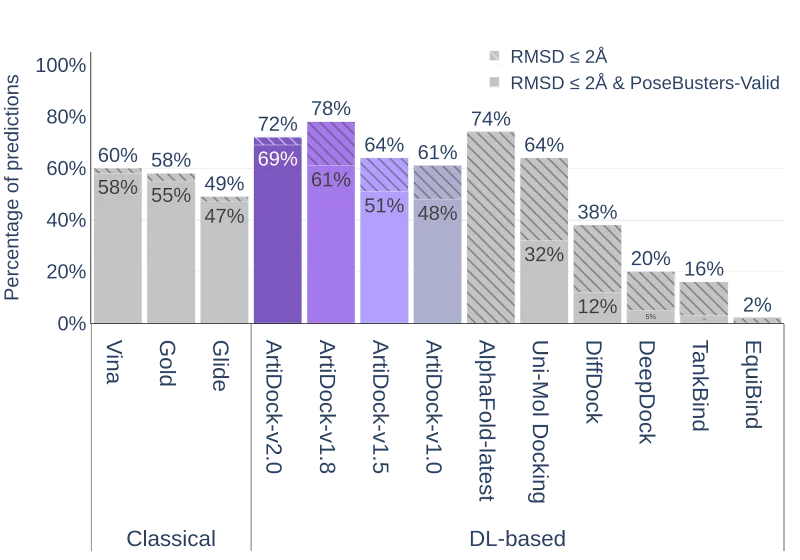
Performance of the docking methods on the PoseBusters v3 dataset (except AlphaFold-Latest, for which only PoseBusters v1 scores are reported in the literature).
It is evident that all ML docking techniques, including ArtiDock up to v1.8, demonstrate a significant gap between the RMSD of all predicted poses and the RMSD of the poses that pass the PoseBusters set of additional quality metrics. For ArtiDock v1.8 this gap is ~17%, which is substantially less than for UniMol (~32%) and DiffDock (~26%), but still a lot more than for classical docking methods (2-3% for Gold, Vina and Glide). This indicates that a considerable fraction of predicted ligand poses in ML docking techniques is still inferior in terms of structural quality metrics.
This issue could be mitigated almost perfectly by improving the distance matrix-to-pose transformation algorithm. In ArtiDock v2.0, an additional penalization of the steric clashes in the differential evolution algorithm for transforming the ligand-protein distance matrix to the ligand coordinates was introduced. This resulted in the drastic decrease of the RMSD gap from 17% in v1.8 to 3% in v2.0.
Moreover, the quality of the posed according to the PoseBusters metrics has also improved drastically: the minimal interatomic distance violations decreased impressively from ~23% in v1.8 to ~5% in v2.0 and the volume overlap with protein and the violation of distances with organic cofactors almost vanished in v2.0.
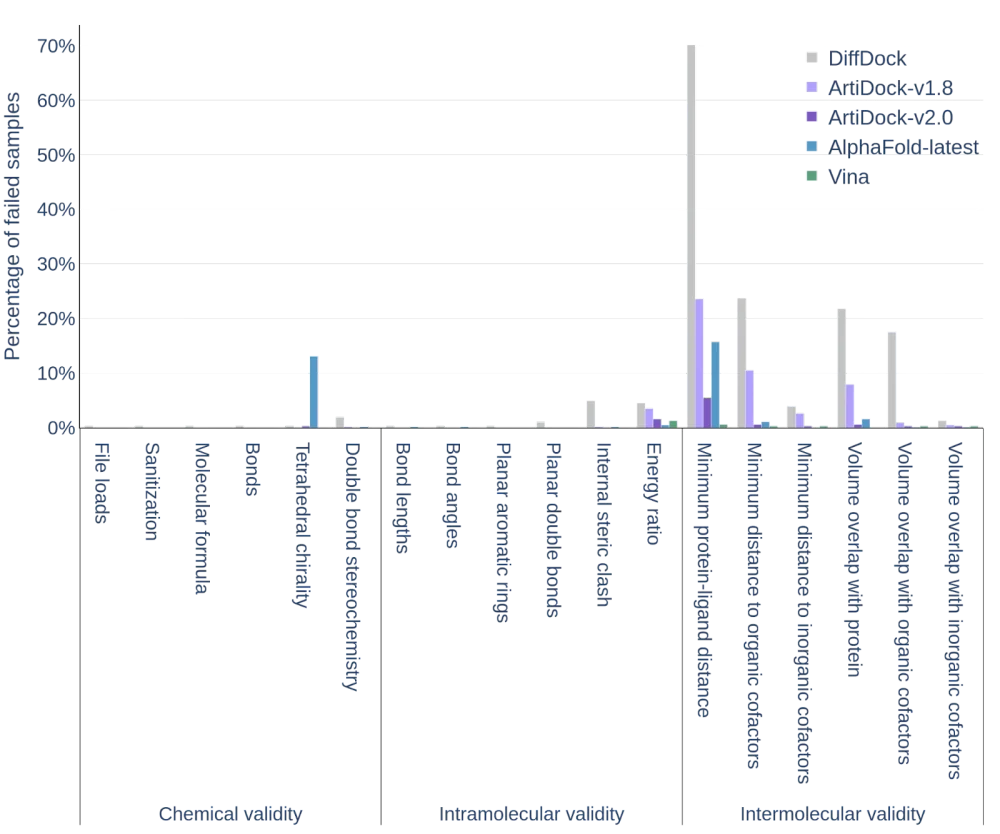
PoseBusters quality metrics of the predicted ligand poses using PoseBusters v3 dataset (except AlphaFold-Latest, for which only PoseBusters v1 scores are reported in the literature). The percentage of failures is reported (the lower, the better).
This impressive improvement in the ligand's poses quality comes at the cost of decreased overall precision in terms of RMSD<2.0Å from ~78% in v1.8 to ~72% in v2.0. However, the percentage of correctly predicted poses with RMSD<2.0Å that are simultaneously PoseBusters-valid increases from 61% to 69%.
Even more important is that this superior performance comes with no inference speed trade-off. ArtiDock 2.0 is at least one order of magnitude faster than conventional docking programs, at least two orders of magnitude faster than DiffDock, and three orders of magnitude faster than AlphaFold-Latest. Combined with RMSD being superior to and the ligand pose quality approaching one of the conventional dockings, this makes this technique an attractive choice for high-throughput applications in real-world drug discovery scenarios.
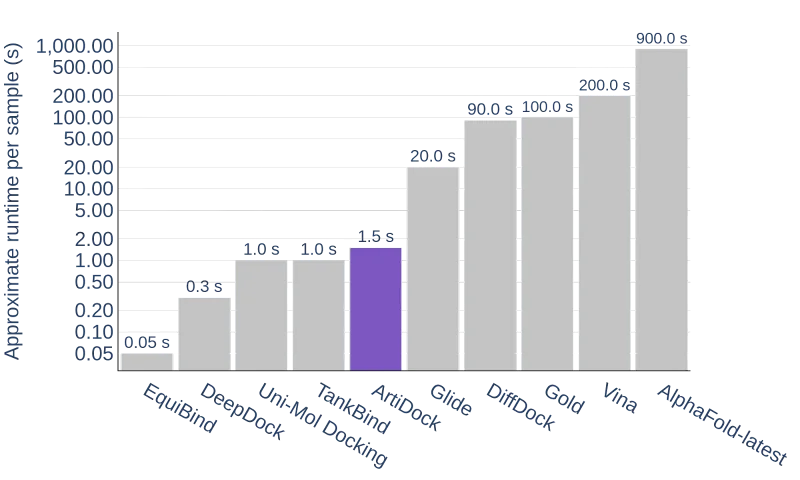
Average binding pose inference time of the docking methods. Note the log scale of the vertical axis.
ArtiDock v2.0 is currently integrated into the production virtual screening pipeline of the Receptor.AI drug discovery platform and already provides impressive results in a series of hit discovery projects. In some of the case studies communicated by Receptor.AI usage of ArtiDock allowed for hit rates of a whopping 40% and the discovery of lead-like compounds with in vivo activity from a single iteration of virtual screening. All this power is available to the customers of Receptor.AI.
ArtiDock is also being prepared to be deployed on the Nvidia BioNeMo cloud platform for drug development, which will make it available to a wide range of interested biotech and pharma companies.